- Products
- CONFLEX
CONFLEX permits fast, accurate, automated conformation searching and analysis critical to drug discovery and chemical engineering. Unique to CONFLEX is its capability to completely search the conformational space of a flexible molecule to find every optimal structure of chemically significant conformers. CONFLEX is not limited to finding only locally-optimized structures that depend on an initial structure input by the user.
Major functions of CONFLEX

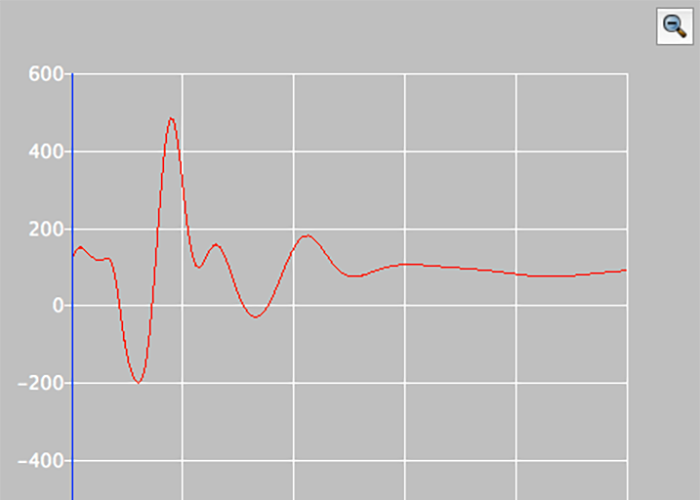
Conformational Space Search
CONFLEX recognizes the molecular structure automatically and identifies a ring and side-chain structures. Then CONFLEX applies three local perturbation methods to generate new conformations: Corner Flap and Edge Flip for the ring structure, Stepwise Rotation for the side chains. These all new structures are geometry optimized and store them to the database.
One of the goal of conformation searching is to find global minimum structure. But in the case of the molecular structure is very flexible, huge number of conformers would be generated. For that reason, CONFLEX employs the reservoir algorithm to generate a new structure from a stable structure at all time, and a range of search limit to search conformers. With these algorithm and limitation, CONFLEX can avoid an explosive growth of conformation number.
Normal vibrational analysis
CONFLEX conducts a normal mode analysis after a geometry optimization automatically. At the same time, CONFLEX calculates a thermodynamic quantities like a Gibbs free energy, etc...
CONFLEX can constrain partially when geometry optimization.
Crystal structure calculation and search
CONFLEX can generate crystal structures automatically from a molecular structure and symmetrical operations defined by specified space groups. CONFLEX optimizes the crystal structures and search minimum energy structures exhaustively. Also, CONFLEX can rank the crystal structures according to their crystal energies or simulated powder X-ray diffraction data.
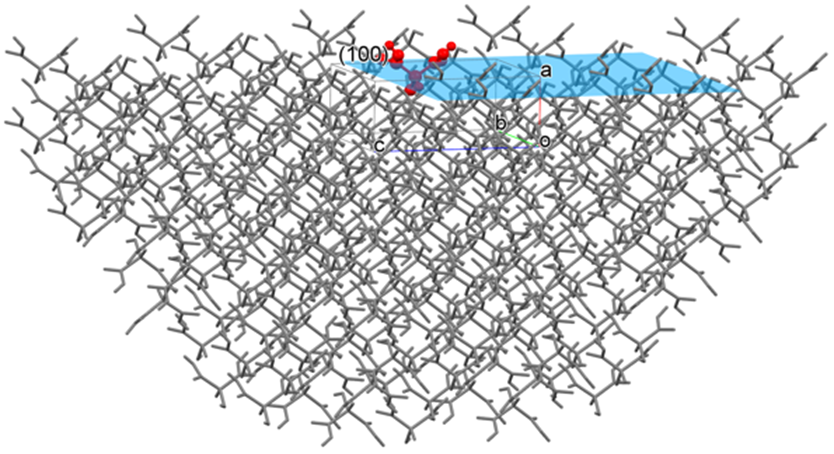
Crystal Surface Analysis
Analysis of crystal surface is essential to understand phenomena of a solid such as dissolution, sublimation, melting and so on.
CONFLEX can calculate a sum of interatomic interaction energies with respect to a molecule in or on a specified crystal plane and evaluate a stability of its plane.
For example, if you specify the (100) plane, CONFLEX builds a hemi-spherical crystal in which the (100) plane is exposed (refer to the below figure) and calculate the sum of interatomic interaction energies with respect to the molecule in or on the (100) plane. A radius of the hemi-spherical crystal is cutoff value.

Solvent effect
GB/SA calculation is applicable to geometry optimization, vibrational analysis and conformational search.
In addition, a logP value is calculated automatically.
Work with Gaussian
If Gaussian program is installed to a computer with CONFLEX program, CONFLEX can invoke Gaussian to optimize and search conformers of a molecule.
With this feature, CONFLEX can handle molecule lacking force field parameter(s) and electronic states which cannot treat by the molecular force field.
Dynamic Reaction Coordinate (DRC)
DRC is molecular dynamics calculation method using initial velocity vectors calculated from normal vibrational modes.
This feature is applicable to configuration changes between multiple molecules or conformation changes of a large molecule.
Host-Ligand Coordination search
CONFLEX had the feature named Host-Ligand coordination search. This is used for specifying energetically stable configurations of complex or molecular cluster.
CD/UV/Vis Spectrum
CONFLEX can simulate CD/UV/Vis spectrum with conformers.
NMR coupling constant analysis
NMR analysis is one of the indispensable measurement methods for analyzing the molecular structures of organic compounds.
Especially for vicinal coupling constant 3J, which depends on the dihedral angle around the center bond (between atoms at the positions 2 and 3), it can be used for the prediction of the three-dimensional structure of the organic compounds by setting the appropriate formulas and parameters. Recently, it has become possible to predict the three-dimensional structures of proteins and other biopolymers by combining the torsion angle based on 3J coupling constant and the predicted distances between proximate protons based on NMR-NOE information. On the other hand, the number of conformational isomers to be considered is increased with increasing the size of molecule, so the measured coupling constants are observed as the average value of the mixture of conformational isomers. As a result, relying solely on measurements enhance the possibility of predicting an “unnatural” mean three-dimensional structure which does not actually exist.
CONFLEX has two types of 3J calculations.
- 3JHH around Csp3-Csp3 bond using Karplus-Imai equation
- 3J for arbitrary combination of four atoms using Karplus equation with user-defined parameters
Furthermore, it is also possible to calculate the 3J values for each conformer generated by the conformational search, and calculate their thermodynamic average. By comparing the calculated values with the measured ones, the state of the molecule observed in NMR analysis can be inferred. In addition, a variety of conformational analyses such as measuring the steric effect on the physical properties and the reactivity and more accurate three-dimensional structure analysis can be supported.
Parameter Configuration
User can customize force field parameter(s) for molecule lacking parameter(s) or modify the existing parameter(s).
The user-customized parameters are only for MMFF94s parameter set.
Amino acid substitution
Some amino acid residues of proteins present in the body may be mutated due to some influence. Even the protein with mutations of only a few residues can have dramatic changes in function and activity. In order to study such the phenomena by a simulation, using pdb file of a protein which has amino acid sequence and can be got from Protein Data Bank (PDB), you should apply amino acid substitutions to the protein for making a mutated protein.
When you use the pdb file provided by the PDB, you may need to correct data in the file due to lack of data on side chains of amino acids. CONFLEX allows you to replace or supplement the lacking side chains of amino acids in the pdb file before starting structure optimization or other calculations. Furthermore, CONFLEX provides you information on the lacking data in the pdb file regardless of side chain and back bone.