Crystal Structure Search
In this section, we will use the "tartronicacid.mol" file in the following folder as an example:
CONFLEX/Sample_Files/CONFLEX/crystal/search
Please copy this file to an appropriate location under your home directory and work on it.
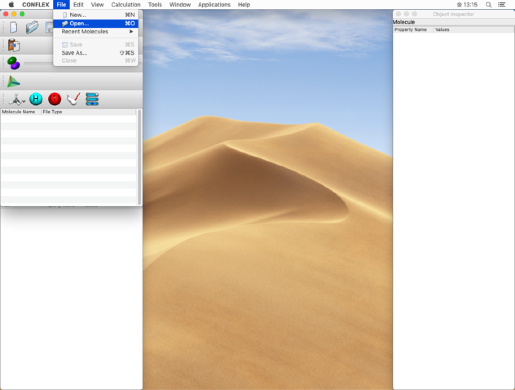
- Click "Open" from the "File" menu to open the copied "tartronicacid.mol" file.
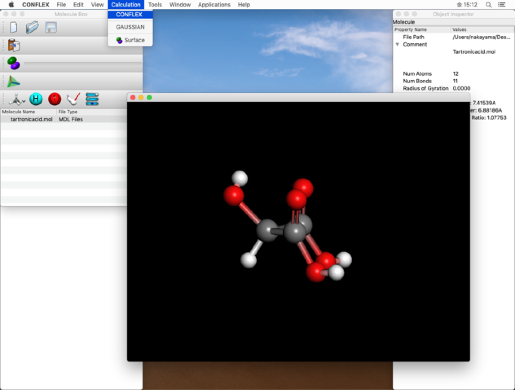
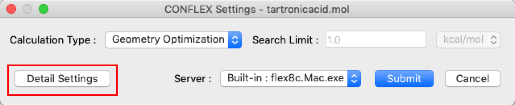
- Select "CONFLEX" from the "Calculation" menu to display the CONFLEX Settings dialog.
- Click "Detail Settings" in the CONFLEX Settings dialog to display the Detail Settings dialog.
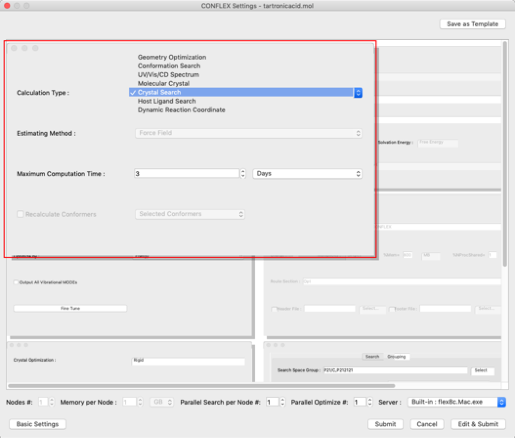
- First, set the type of calculation.
- Next, set the optimization method.
- Next, set the search method in the "Crystal Search" dialog at the bottom right.
- Rotation Method:
- Grid
- Rotation Step:
- 30.00
- Position Prediction Method:
- Random
- Trial Structures:
- 20
- Click "Submit" button in the Detail settings dialog.
- When the job is started, the "Job Manager" will be displayed.
-
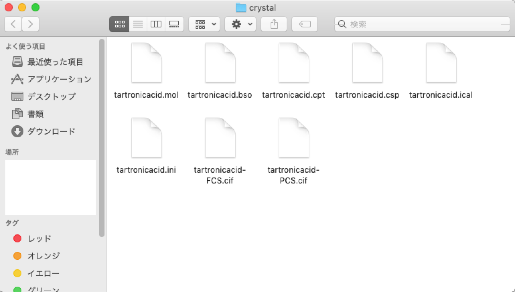
The folder contains "tartronicacid.mol" and seven other files:
- tartronicacid.bso
- tartronicacid.cpt
- tartronicacid.csp
- tartronicacid.ical
- tartronicacid.ini
- tartronicacid-FCS.cif
- tartronicacid-PCS.cif
Select "Crystal Search" from the "Calculation Type:" pull-down menu in the "General Settings" dialog on the top left.
Select "Rigid" from the "Crystal Optimization:" pull-down menu in the "Crystal Optimization" dialog on the lower left.
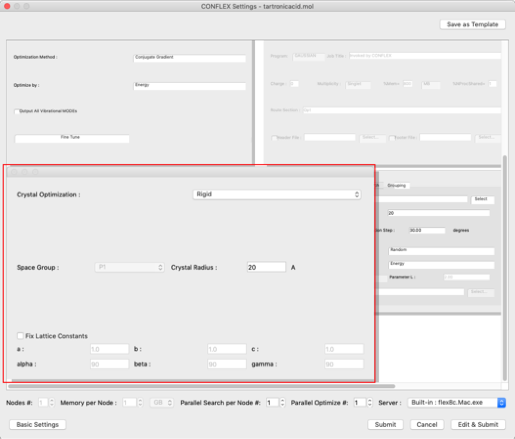
Set each of the following:
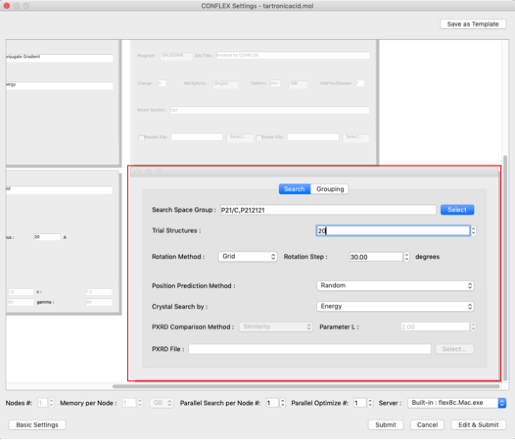
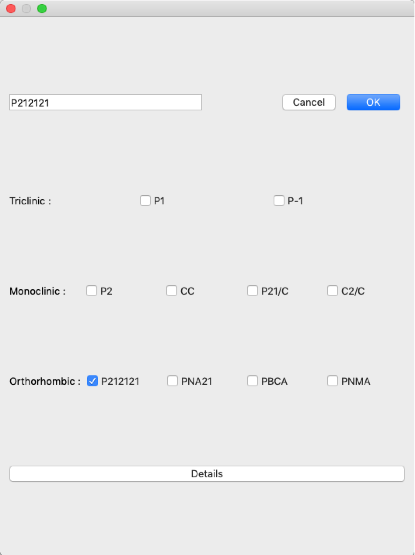
Set the space group of the search to "P212121".
Click "Select" button on the right of "Search Space Group:" and check "P212121" in the dialog that appears.
Make sure all other checkboxes are unchecked and click "OK".
Each setting will affect the search results. For details on each setting, please refer to the manual or tutorial for details on each setting.
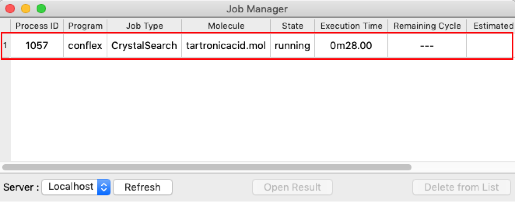
The crystal structure search job will be started.

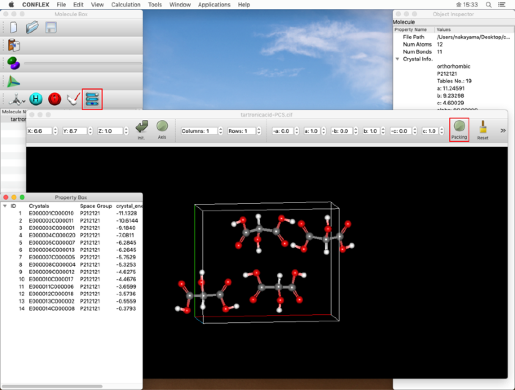
After confirming that the "State" is "Finished", double-click on the row indicated by the red frame.
The crystal structure obtained from the search is displayed in the molecule window that opens by double-clicking. Other crystal structures are listed in the "Property Box" dialog, and by clicking on an energy value, its crystal structure will be displayed in the molecule window.