Gaussian Settings
When [GAUSSIAN] is selected in the [Calculation] menu, the Gaussian basic settings dialog is displayed.
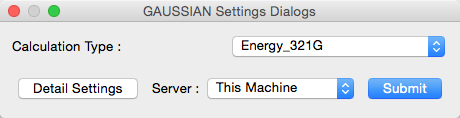
This dialog is used in same way with CONFLEX basic set- ting dialog.
When the [Detail Settings] button is clicked, the detail setting dialog is displayed.
The dialogs are in the form of an embedding of each configuration dialog, allowing you to see the whole picture.
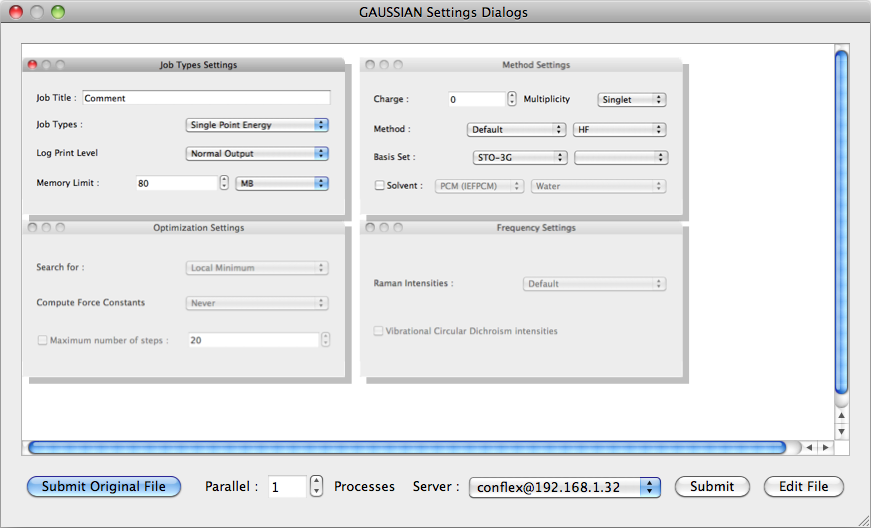
- When the file is opened in the [Gaussian Input] format and when the [Submit Original File] button at the lower left of the dialog is clicked, the original file can be inputted for a job as is.
- To run parallel calculations in a parallel environment, the setting for [Parallel] must be greater than 1. Please note that LINDA cannot be handled in the present version of CONFLEX Interface.
- The location for carrying out calculations is designated in [Server:]
- When the [Submit] button is clicked, the job is submitted to the Job Manager.
- When the [Edit File] button is clicked, the text editor opens and the settings can be edited manually. This would be used to add a setting (keyword) if not present in the dialog box.
Job Types Dialog
The type of job is set using this dialog.
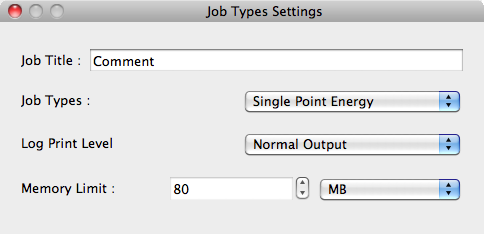
Job Title:
Type your description into the [Job Title] text box.
Job Types:
The type of calculation is selected in [Job Types]. The following typical calculations can be selected:
- Single Point Energy (SP)
- Geometry Optimization (OPT)
- Frequency Analysis (FREQ)
- Optimization + Frequency (OPT FREQ)
- Reaction Path Following (IRC)
- Polarizabilities (POLAR)
- Polar + Frequency (POLAR FREQ)
- Forces on Nuclei (FORCE)
- Wavefunction Stability (STABLE)
- Molecular Volume (VOLUME)
See the Gaussian Manual for details:
List of Gaussian Keywords (An external site will open in a new window.)
Log Print Level
Log file settings in [Log Print Level] are:
- Additional Output
- most detailed output
- Normal Output
- normal output
- Terse Output
- minimum output
Memory Limit:
When running Gaussian jobs, you will need to set the amount of memory used for calculations in [Memory Limit:]. If not enough memory is allocated, the calculations may fail due to insufficient memory. If too much memory is allocated, this may lead to a decline in performance.
The units are selected from the following:
- MB
- MW
- GB
- GW
- TB
- TW
For shared-memory parallel jobs, the amount of allocated memory is need to be increased appropriately to the number of processors/cores used in calculation. Please see the Gaussian Manual for additional details.
Link 0 Commands (An external site will open in a new window.)
Running Gaussian (An external site will open in a new window.)
Method Dialog
Calculation methods are configured here.
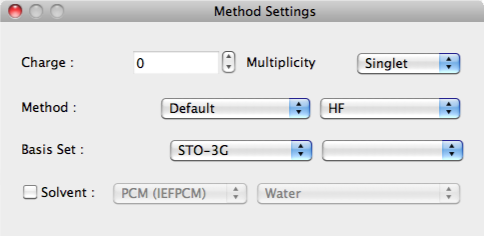
Charge:
The formal charge for the overall molecule is designated in [Charge:].
Multiplicity
The spin multiplicity is designated in [Multiplicity:].
Method, Basis Set
The method of calculation is set in [Method:] and [Basis Set:]. The selection options change depending on the method selected.
The restriction method is selected from the following:
- Default
- Restricted
- restricted method
- Unrestricted
- unrestricted method
- Restricted Open
- open shell method with restrictions
When [Semi Empirical] is used, a choice can be made with the restriction method and a method from the [Basis Set:]:
- PM6
- AM1
- PM3
- ZINDO
The following can be selected in [Basis Set:] in [HF], [DFT] and [MP2].
- STO-3G
- 3-21G
- 6-31G
- 6-311G
- LanL2MB
- LanL2DZ
The diffuse and polarization function are displayed in response to the selected basis set.
Selections can be made from the followings for [DFT]:
- B3LYP
- CAMB3LYP
- B3PW91
- mPW1PW91
Solvent
When the [Solvent] box is ticked, the following selections are available in the adjacent menu:
- PCM (IEFPCM)
- CPCM
- Dipole
- IPCM
- SCIPCM
You can also specify the solvent by selecting from the popup menu. Use the [Edit File] function to specify a solvent other than those on the solvent list. For more detail on the acceptable solvent option, please refer to the Gaussian manual.
Optimization Dialog
Geometry optimization settings are configured here.
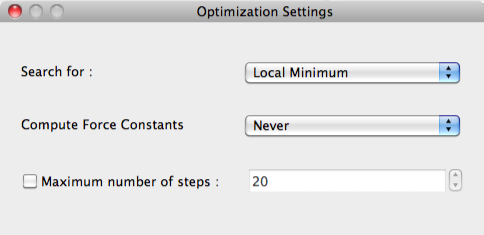
Search for:
Selections can be made for [Local Minimum] or for the [Transition State] in [Search for:]:
Compute Force Constants
The frequency of calculating the constant of force is designated in [Compute Force Constants]:
- Never
- not calculated
- Once
- calculated only once when calculation starts
- Always
- always calculated
Maximum number of steps
The maximum value of iterations for geometry optimization is set in [Maximum number of steps].
Frequency Dialog
In this dialog we can configure the settings for vibrational calculations.
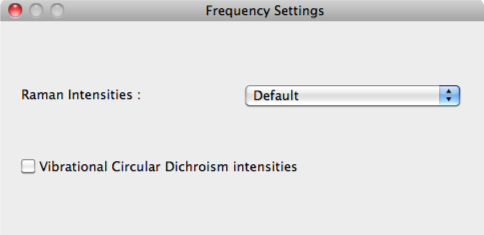
Raman Intensities
A designation is made in [Raman Intensities] whether to carry out calculations for the Raman spectrum.
Vibrational Circular Dichroism Intensities
To run VCD calculations, tick the check box next to [Vibrational Circular Dichroism Intensities].
Array Job Settings
When the MDL SD file is used as input, multiple input files are generated and multiple jobs are submitted at one time.
When an MDL SD file is opened and a calculation is started without selecting any displayed structure in the Property Editor window, all of the structures are in scope. Multiple structures can be selected for calculation by holding down the shift key and selecting them in the list with the mouse click.
When the array job submitted, the Job Name in the Job Manager is modified with the word “array-”. The Array Job execution status is indicated in the [Array Task ID] column. In the example in the figure, the 31st, 32nd, 33rd, 34th, 35th and 36th structures are executed and the 37th through the 59th structures are in standby mode. Calculation of structures 1 through 30 is completed.

If the job is cancelled, all of the calculations, including those in pending mode, are cancelled as well. In the current version, a specific job can not be cancelled if it was submitted in batch.
After the calculations have been completed and results are viewed, the screen will appear the same as when an MDL SD file has been opened. However, the appeared structures are corresponding to the results of calculation.
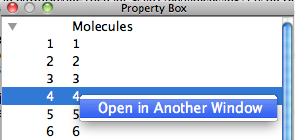
When each structure in the calculation results is opened individually, the list in [Property Box] is right-clicked and [Open in Another Window] can be selected from the context menu displayed.
The filename for each structure in an Array Job is the original file name plus an ID number, (e.g. molecule_0007.log).
If you want to open a folder containing all of the calculation results, right-click the filename in the [Molecule Box] and select [Open Folder] from the context menu.