Conformation search
This section explains how to execute a conformation search using CONFLEX. The “Cyclohexane-F.mol” file obtained in the “Execution of geometry optimization and vibrational analysis” is used as structure file. The Cyclohexane-F.mol contains data of the optimized structure of Cyclohexane.
[Execution of conformation search]
[Execution from Interface]
Open the Cyclohexane-F.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then select [Conformation Search] from the [Calculation Type:] pull-down menu on the calculation setting dialog that appears. Leave the value of [Search Limit:] at 1.0. This parameter is used as a criterion for selecting initial structures in the conformation search. During the calculation, conformers found within 1.0 kcal/mol from the most stable conformer are selected as initial structures. Therefore, when the calculation is complete, the conformation search within 1.0 kcal/mol energy range is considered to be finished.
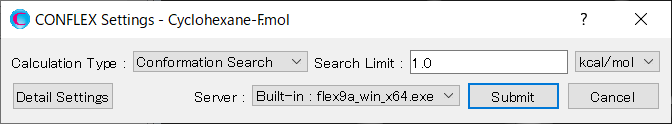
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the Cyclohexane-F.ini file.
Cyclohexane-F.ini file
MMFF94S CONFLEX SEARCH=ENERGY SEL=1.0
Explanations of each keyword are shown below.
- MMFF94S
- Use MMFF94s force field
- CONFLEX
- Execute the conformation search
- SEARCH=ENERGY
- Search limit on conformational space is determined by energy.
- SEL=1.0
- The search limit sets to 1.0 kcal/mol.
The search limit is used as a criterion for selecting initial structures in the conformation search. During the calculation, conformers found within 1.0 kcal/mol from the most stable conformer are selected as initial structures. Therefore, when the calculation is complete, the conformation search within 1.0 kcal/mol energy range is considered to be finished.
The settings specified by the keywords “MMFF94S”, “SEARCH=ENERGY”, and “SEL=1.0” are equivalent to the default settings. Therefore, even if you use the following ini file, the settings in both calculations will be the same.
Cyclohexane-F.ini file
CONFLEX
Store the two files of Cyclohexane-F.mol and Cyclohexane-F.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Cyclohexane-Fenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
[Output files]
Three files, -F.mol, sdf, and bso files, namely Cyclohexane-F-F.mol, Cyclohexane-F.sdf, and Cyclohexane-F.bso are output just as in the case of the geometry optimization calculation. Additionally, in the case of the conformation search, the following files are also generated.
- Cyclohexane-F.fxf
- This file contains the structure data of conformers found during the conformation search. If you restart the conformation search using this file, you will only explore an extended conformational space from the previous results.
- Cyclohexane-F.fxo
- This file contains detailed information on the conformation search such as the local perturbations by flip-flap and corner flap and the relationships between the conformers already identified during the search.
- Cyclohexane-F.ls1
- The conformers found by the search are listed in order of steric energy in this file.
- Cyclohexane-F.ls2
- This file lists the dihedral angle values used to identify duplicate conformers during the search.
- Cyclohexane-F.ls3
- This file contains data for each conformer found in the search, including the CONF. ID, steric energy, distribution based on steric energy, atomic coordinates, and atom list.
- Cyclohexane-F.ls4
- The conformers identified through the search are listed in this file according to free energy.
- Cyclohexane-F.sdf
-
The structure data of each conformer found by the search are stored in this file in MDL-MOL format. An explanation of the MDL-MOL format is provided in the Explanation about Cyclohexane.mol file.
While the MOL file contains data for a single structure, the SDF file can include multiple structure data entries, separated by “$$$$”. Therefore, by dividing the structure data in the SDF file using “$$$$”, you can obtain the MOL file for each individual structure. The structure data in the SDF file are ordered according to total Gibbs free energy. Each structure data entry is identified by “IFN=”, where the first number represents the Gibbs free energy rank and the second number is the unique conformer ID assigned during the search.
0.7343750
18
2
1.00000000000000
5.200
1
T
F
0
F
6
D3D
-3.56093559779315
3.249848646468316E-009
0.888728442903559 1.48121407150593 39.2016730029237
-10.2067647343158 4.96801633911096 0.888728442903559
0.888728442903559 22.7814526607543 -5.90356166790034
2.98080980346657 106.763590387419 106.763590387419
8.44981392719261 104.244278365026 16.4665174320397
-3.56093559779315 -3.56093559779315 0.000000000000000E+000
-3.56093559779315 0.000000000000000E+000 0.000000000000000E+000
0.000000000000000E+000 0.000000000000000E+000 0.000000000000000E+000
0.000000000000000E+000 104.980111675433 105.572597304035
70.4329395908707 84.5730163650170 24.4153435746172
105.050542904982 13.6944155520592 0.000000000000000E+000
56.7385240388114 6.00000000000000
116.679654042328 116.679654042695 204.596311457385
0.000000000000000E+000 0.000000000000000E+000
***** START LOOP OF CONFLEX SEARCH ***** 18 TRIAL STRUCUTRES GENERATED FROM 1 INITIAL STRUCTURE(S). COUNT OF STACKED INITIAL STRUCUTRES: 1 INITIAL STRUCTURE - IDN: 1, EGY: -3.5609 NUMBER OF TRIAL CONFORMATIONS GENERATED: 18 NUMBER OF INITIAL STRUCTURES LEFT: 1 A NEW CONFORMER - IFT: 21,IDN: 2, EGY: 2.3688, GRMS: 0.81E-07, TIME: 0.05 REPLACED OLD ONE - IFT: 22,IDN: 2, EGY: 2.3688, GRMS: 0.50E-08, TIME: 0.03 IDENTICAL OLD ONE - IFT: 23,IDN: 2, EGY: 2.3688, GRMS: 0.74E-07, TIME: 0.03 REPLACED OLD ONE - IFT: 24,IDN: 2, EGY: 2.3688, GRMS: 0.98E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 25,IDN: 2, EGY: 2.3688, GRMS: 0.64E-06, TIME: 0.03 IDENTICAL OLD ONE - IFT: 26,IDN: 2, EGY: 2.3688, GRMS: 0.17E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.14E-06, TIME: 0.03 REPLACED OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.32E-08, TIME: 0.03 REPLACED OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.20E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.14E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.73E-07, TIME: 0.03 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.14E-07, TIME: 0.05 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.17E-08, TIME: 0.03 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.11E-06, TIME: 0.03 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.37E-08, TIME: 0.03 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY: -3.5609, GRMS: 0.15E-08, TIME: 0.08 IDENTICAL OLD ONE - IFT: 10,IDN: 2, EGY: 2.3688, GRMS: 0.26E-09, TIME: 0.03 IDENTICAL OLD ONE - IFT: 20,IDN: 1, EGY:
!====================================================================================!
! !
! LIST OF CONFORMERS (LIST FORM 1) !
! !
!------------------------------------------------------------------------------------!
! !
! DATE: 2020/09/04 TIME: 16:04:19.46 !
! Cyclohexane-F: Cyclohexane.mol !
! EMPIRICAL FORMULA: C6H12 MW = 84.094 !
! FORCE FIELD: MMFF94S(2010-12-04HG) !
! TOTAL NUMBER OF CONFORMERS FOUND: 2 !
! STERIC ENERGY: MIN= -3.5609 MAX= 2.3688 AVERAGE= -3.5607 !
! !
!------------------------------------------------------------------------------------!
! !
! DEFINITIONS: !
! NO. = INDEX NUMBER IN THE ORDER OF STERIC ENERGY !
! CONF. ID = IDENTIFICATION NUMBER OF EACH CONFORMER !
! STERIC E = STERIC ENERGY OF EACH CONFORMER (KCAL/MOL) !
! DELTA E = RELATIVE STERIC ENERGY FROM THE GLOBAL ENERGY MINIMUM (KCAL/MOL) !
! DISTRIBUTION = DISTRIBUTION BASED ON STERIC ENERGY (%) !
! INIT. = FLAG OF THE INITIAL STRUCTURE THAT HAD BEEN ALREADY USED !
! "*" = INDICATES THE INITIAL STRUCTURE USED IN THIS JOB !
! REOPT. = FLAG OF THE OPTIMIZED STRUCTURE THAT HAD BEEN OPTIMIZED AGAIN !
! "+" = INDICATES THE NEW CONFORMERS FOUND IN THIS JOB !
! NO. NEG. = NUMBER OF NEGATIVE EIGEN VALUES OF EACH CONFORMER !
! !
!====================================================================================!
====================================================================================
CONF. ORIGINAL DISTRI- NO.
NO. ID STERIC E DELTA E BUTION INIT. REOPT. NEG.
------------------------------------------------------------------------------------
1 00000001 -3.5609 0.0000 99.9955 T* F+ 0
2 00000002 2.3688 5.9297 0.0045 F F+ 0
------------------------------------------------------------------------------------
MINIMUM ENERGY: -3.5609 KCAL/MOL
AVERAGE ENERGY: -3.5607 KCAL/MOL (BASED ON THE BOLTZMANN LAW)
====================================================================================
Explanations of each item are provided below.
| Item | Explanation |
|---|---|
| NO. | Number ordered by steric energy |
| CONF.ID | Number assigned based on the order found by the search |
| ORIGINAL STERIC E | Steric energy (kcal/mol) |
| DELTA E | Relative steric energy from the lowest one (kcal/mol) |
| DISTRIBUTION | Conformation distribution obtained using Boltzmann distribution based on the steric energy |
| INIT. | Flag indicating whether it was used as an initial structure |
| REOPT. | Flag indicating whether the structure has already been optimized or whether the geometry optimization was performed in current conformer search calculation. |
| NO. NEG. | The number of negative eigenvalues obtained from normal mode analysis |
In the value of INIT., the “T” indicates that the conformer has already been used as an initial structure, while “*” means that the conformer was used as an initial structure in the current conformational search calculation.
The status of geometry optimization during conformational search is indicated by “F” and “+”. The “F” means that the structure has already been optimized, while “+” signifies that the structure is a newly found conformer in the current conformational search calculation.
Regarding “NO. NEG”, the value of "0" indicates that all eigenvalues are positive in the normal mode analysis of the conformer. If the conformer has any negative eigenvalues, the number of such values is displayed. When one negative eigenvalue is present, the structure is considered a transition state connecting equilibrium conformers. To more accurately confirm whether it is a transition state, it is recommended to perform geometry optimization, normal mode analysis, and intrinsic reaction coordinate calculation using an ab initio method.
!------------------------------------------------------------------------------------!
! !
! DEFINITIONS OF TORSION ANGLES: !
! D 1 = 3 - 1 - 2 - 16 !
! D 2 = 1 - 2 - 16 - 13 !
! D 3 = 2 - 16 - 13 - 8 !
! D 4 = 16 - 13 - 8 - 3 !
! D 5 = 13 - 8 - 3 - 1 !
! D 6 = 8 - 3 - 1 - 2 !
! !
!====================================================================================!
============================================================================================================
NO. ID STERIC E DELTA E DISTRIB. D 1 D 2 D 3 D 4 D 5 D 6
------------------------------------------------------------------------------------------------------------
1 00000001 -3.5609 0.0000 99.9955 -54.26 54.26 -54.26 54.26 -54.26 54.26
2 00000002 2.3688 5.9297 0.0045 29.17 29.17 -60.11 29.17 29.17 -60.11
------------------------------------------------------------------------------------------------------------
AVERAGE: -3.5607 0.0003
============================================================================================================
00000001 E= -3.5609; P= 99.9955 18 120. -1.26267 0.72901 0.22578 1 -0.00000 1.45801 -0.22578 1 -1.26267 -0.72901 -0.22578 1 -2.14619 1.23910 -0.17419 5 -1.33651 0.77163 1.31946 5 0.00000 2.47821 0.17419 5 -0.00000 1.54327 -1.31946 5 -0.00000 -1.45801 0.22578 1 -2.14619 -1.23910 0.17419 5 -1.33651 -0.77163 -1.31946 5 -0.00000 -2.47821 -0.17419 5 -0.00000 -1.54327 1.31946 5 1.26267 -0.72901 -0.22578 1 2.14619 -1.23910 0.17419 5 1.33651 -0.77163 -1.31946 5 1.26267 0.72901 0.22578 1 1.33651 0.77163 1.31946 5 2.14619 1.23910 -0.17419 5 STERIC E.= -3.56094 T F 00000002 E= 2.3688; P= 0.0045 18 120. -0.67151 1.22910 0.35868 1 -1.50939 0.00000 0.00000 1 0.67151 1.22910 -0.35868 1 -0.49926 1.24725 1.44233 5 -1.23203 2.13933 0.11739 5 -2.16866 0.23360 -0.84486 5 -2.16866 -0.23360 0.84486 5 1.50939 -0.00000 0.00000 1 0.49926 1.24725 -1.44233 5 1.23203 2.13933 -0.11739 5 2.16866 0.23360 0.84486 5 2.16866 -0.23360 -0.84486 5 0.67151 -1.22910 0.35868 1 1.23203 -2.13933 0.11739 5 0.49926 -1.24725 1.44233 5 -0.67151 -1.22910 -0.35868 1 -0.49926 -1.24725 -1.44233 5 -1.23203 -2.13933 -0.11739 5 STERIC E.= 2.36879 F F
!====================================================================================!
! !
! LIST OF CONFORMERS (LIST FORM 4) !
! !
!------------------------------------------------------------------------------------!
! !
! DATE: 2020/09/04 TIME: 16:04:19.46 !
! Cyclohexane-F: Cyclohexane.mol !
! EMPIRICAL FORMULA: C6H12 MW = 84.094 !
! FORCE FIELD: MMFF94S(2010-12-04HG) !
! TOTAL NUMBER OF CONFORMERS FOUND: 2 !
! STERIC ENERGY: EMIN= -3.5609 EMAX= 2.3688 EAVE= -3.5607 !
! GIBB'S FREE ENERGY: GMIN= 84.5730 GMAX= 88.7989 GAVE= 84.5764 !
! MIXING ENERGIES: HMIX= 105.5770 TSMIX= 21.0044 GMIX= 84.5725 !
! MIXING HEAT CAPACITY: CPMIX= 24.4158 !
! TEMPERATURE: 25.00 CELSIUS ( 298.15 KELVIN) !
! !
!------------------------------------------------------------------------------------!
! !
! DEFINITIONS: !
! NO. = INDEX NUMBER IN THE ORDER OF THE TOTAL GIBBS'S FREE ENERGY !
! CONF. ID = IDENTIFICATION NUMBER OF EACH CONFORMER !
! HE = ENTHALPY CONTRIBUTION OF EACH CONFORMER (KCAL/MOL) !
! TSE = ENTROPY (MULRPLIED BY TEMP.) CONTRIBUTION OF EACH CONFORMER (KCAL/MOL)!
! GE = TOTAL GIBB'S FREE ENERGY OF EACH CONFORMER (KCAL/MOL) !
! DGE = RELATIVE GIBB'S FREE ENERGY OF EACH CONFORMER (KCAL/MOL) !
! GEPOP = DISTRIBUTION IN GIBB'S FREE ENERGY (%) !
! SE = STERIC ENERGY OF EACH CONFORMER (KCAL/MOL) !
! DSE = RELATIVE STERIC ENERGY FROM THE GLOBAL ENERGY MINIMUM (KCAL/MOL) !
! SEPOP = DISTRIBUTION BASED ON STERIC ENERGY (%) !
! CHIRAL = CHIRALITY EXISTANCE OF EACH CONFORMER !
! SYM. NUM. = SYMMETRY NUMBER OF EACH CONFORMER !
! POINT GROUP = POINT GROUP OF SYMMETRY OF EACH CONFORMER !
! INIT. = FLAG OF EACH CONFORMER THAT HAD USED FOR INITIAL STRUCTURE !
! REOPT. = FLAG OF EACH CONFORMER THAT HAD BEEN OPTIMIZED AGAIN !
! NO. NEG. = NUMBER OF NEGATIVE EIGEN VALUES OF EACH CONFORMER !
! !
!====================================================================================!
========================================================================================================================================
CONF. SYM. POINT NO.
NO. ID HE TSE GE DGE GPOP SE DSE EPOP CHIRAL NUM. GROUP INIT. REOPT. NEG.
----------------------------------------------------------------------------------------------------------------------------------------
1 00000001 105.5726 20.9996 84.5730 0.0000 99.9202 -3.5609 0.0000 99.9955 F 6 D3D T F 0
2 00000002 111.0426 22.2436 88.7989 4.2259 0.0798 2.3688 5.9297 0.0045 T 4 D2 F F 0
----------------------------------------------------------------------------------------------------------------------------------------
========================================================================================================================================
Explanations of each item are shown below.
| Item | Explanation |
|---|---|
| NO. | Number ordered by total Gibbs free energy |
| CONF.ID | Number assigned based on the order found by the search |
| HE | Enthalpy (kcal/mol) |
| TSE | Entropy multiplied by temperature (kcal/mol) |
| GE | Total Gibbs free energy (kcal/mol) |
| DGE | Relative Gibbs free energy form the lowest one (kcal/mol) |
| GPOP | Conformation distribution obtained using Boltzmann distribution based on Gibbs free energy |
| SE | Steric energy (kcal/mol) |
| DSE | Relative steric energy from the lowest one (kcal/mol) |
| EPOP | Conformation distribution obtained using Boltzmann distribution based on steric energy |
| CHIRAL | Flag indicating whether the conformer has chirality. |
| SYM. NUM. | Symmetry number |
| POINT GROUP | Symbol of point group |
| INIT. | Flag indicating whether it was used as an initial structure |
| REOPT. | Flag indicating whether the structure has already been optimized or whether the geometry optimization was performed in current conformer search calculation. |
| NO. NEG. | The number of negative eigenvalues obtained from normal mode analysis |
Cyclohexane.mol
CONFLEX 20090416043D 1 1.00000 -3.56094 1
D3D ,E = -3.561, G = 0.325E-08, P = 99.9955, M( 0), IFN =00000001-00000001
18 18 0 0 999 V2000
-1.2627 0.7290 0.2258 C 0 0 0 0 0
-0.0000 1.4580 -0.2258 C 0 0 0 0 0
-1.2627 -0.7290 -0.2258 C 0 0 0 0 0
-2.1462 1.2391 -0.1742 H 0 0 0 0 0
-1.3365 0.7716 1.3195 H 0 0 0 0 0
0.0000 2.4782 0.1742 H 0 0 0 0 0
-0.0000 1.5433 -1.3195 H 0 0 0 0 0
-0.0000 -1.4580 0.2258 C 0 0 0 0 0
-2.1462 -1.2391 0.1742 H 0 0 0 0 0
-1.3365 -0.7716 -1.3195 H 0 0 0 0 0
-0.0000 -2.4782 -0.1742 H 0 0 0 0 0
-0.0000 -1.5433 1.3195 H 0 0 0 0 0
1.2627 -0.7290 -0.2258 C 0 0 0 0 0
2.1462 -1.2391 0.1742 H 0 0 0 0 0
1.3365 -0.7716 -1.3195 H 0 0 0 0 0
1.2627 0.7290 0.2258 C 0 0 0 0 0
1.3365 0.7716 1.3195 H 0 0 0 0 0
2.1462 1.2391 -0.1742 H 0 0 0 0 0
2 1 1 0 0
3 1 1 0 0
1 4 1 0 0
1 5 1 0 0
2 6 1 0 0
2 7 1 0 0
2 16 1 0 0
3 8 1 0 0
3 9 1 0 0
3 10 1 0 0
11 8 1 0 0
12 8 1 0 0
13 8 1 0 0
14 13 1 0 0
13 15 1 0 0
16 13 1 0 0
16 17 1 0 0
16 18 1 0 0
M END
> <DATE_YYYY/MM/DD>
2020/09/04
> <TIME_HH:MM:SS.XX>
16:04:19.46
> <MOL_FILE_NAME>
Cyclohexane-F: Cyclohexane.mol
> <FORCE_FIELD_NAME>
MMFF94S(2010-12-04HG)
> <TEMPERATURE_K>
298.15
> <TOTAL_NUMBER_OF_CONFORMERS>
2
> <CONFORMER_ID>
00000001
> <POTENTIAL_ENERGY_KCAL/MOL>
-3.560936
> <ENERGY_RMS_GRADIENT_KCAL/MOL/ANGS>
3.2500000E-09
> <BOLTZMANN_POPULATION_%>
99.99550
> <TOTAL_GIBBS_FREE_ENERGY_KCAL/MOL>
84.57302
> <TOTAL_ENTHALPY_KCAL/MOL>
105.5726
> <TOTAL_ENTROPY_CAL/MOL/K>
70.43294
> <HEAT_CAPACITY_CAL/MOL/K>
24.41534
> <ENTHALPY_FUNCTION_CAL/MOL/K>
13.69442
> <FREE_ENERGY_FUNCTION_CAL/MOL/K>
56.73852
> <CHIRALITY>
F
> <SYMMETRY_NUMBER>
6
> <POINT_GROUP>
D3D
> <NUMBER_OF_NEGATIVE_EIGENVALUES>
0
> <CONFLEX_PERTURBATION_FLAG>
T
> <CONFLEX_REOPTIMIZATION_FLAG>
F
$$$$
[Expansion of conformation search space]
Refer to Cyclohexane-F.ls1 in the previous section. The second conformer shows “F” in “INIT.” column, indicating that it has not yet been used as an initial structure in the search. This suggests that new conformers may be discovered using the second conformer as a starting point. Therefore, we restart the conformational search using the fxf file and perform an expanded search with the second conformer as the initial structure.
[Execution from Interface]
Open Cyclohexane-F.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then select [Conformation Search] from the [Calculation Type:] pull-down menu on the calculation setting dialog that appears.
Edit the value of [Search Limit:] to 6.0. This is because the energy difference between the most stable conformer and the second most stable conformer is 5.9297 kcal/mol. With this setting, the second most stable conformer will be included as initial structure in the search.
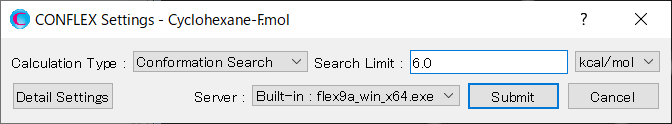
After completing the calculation settings, click to start the calculation.
After finishing this calculation, the search of the conformational space within 6.0 kcal/mol of the most stable conformer is completed.
For the expanded search calculation, the Cyclohexane-F.fxf file obtained from the previous search is required in the folder containing the Cyclohexane-F.mol and Cyclohexane-F.ini files.
[Execution from command line]
Edit the value of [SEL=] to 6.0 in Cyclohexane-F.ini file.
This is because the energy difference between the most stable conformer and the second most stable conformer is 5.9297 kcal/mol. With this setting, the second most stable conformer will be included as initial structure in the search.
Cyclohexane-F.ini file
MMFF94S CONFLEX SEARCH=ENERGY SEL=6.0
Store the three files of Cyclohexane-F.mol, Cyclohexane-F.ini, and Cyclohexane-F.fxf in a single folder, and execute the following command to start the calculation.
After finishing this calculation, the search of the conformational space within 6.0 kcal/mol of the most stable conformer is completed.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Cyclohexane-Fenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
If you check Cyclohexane-F.ls1, you will see that the value of "INIT." for the second conformer has changed to “T”. This indicates that the second conformer was used as an initial structure in the search.
====================================================================================
CONF. ORIGINAL DISTRI- NO.
NO. ID STERIC E DELTA E BUTION INIT. REOPT. NEG.
------------------------------------------------------------------------------------
1 00000001 -3.5609 0.0000 99.9955 T F 0
2 00000002 2.3688 5.9297 0.0045 T* F 0
------------------------------------------------------------------------------------
MINIMUM ENERGY: -3.5609 KCAL/MOL
AVERAGE ENERGY: -3.5607 KCAL/MOL (BASED ON THE BOLTZMANN LAW)
====================================================================================
New conformers were not obtained in the expanded search. Therefore, it can be concluded that all minimum energy structures accessible using the CONFLEX algorithm and the MMFF94s force field have been identified.
The region of conformational space to be explored can be expanded by increasing the value of SEL, based on the information in the ls1 file, and re-running the search. When all conformers found are used as initial structures and no new conformers are generated, it can be considered that the conformational space defined by the applied force field has been fully explored.
[Visualization of calculation results]
[If you executed the calculation using Interface]
After submitting a job, the Job Manager appears at the bottom of the CONFLEX interface. The Job Manager displays the status of the job.
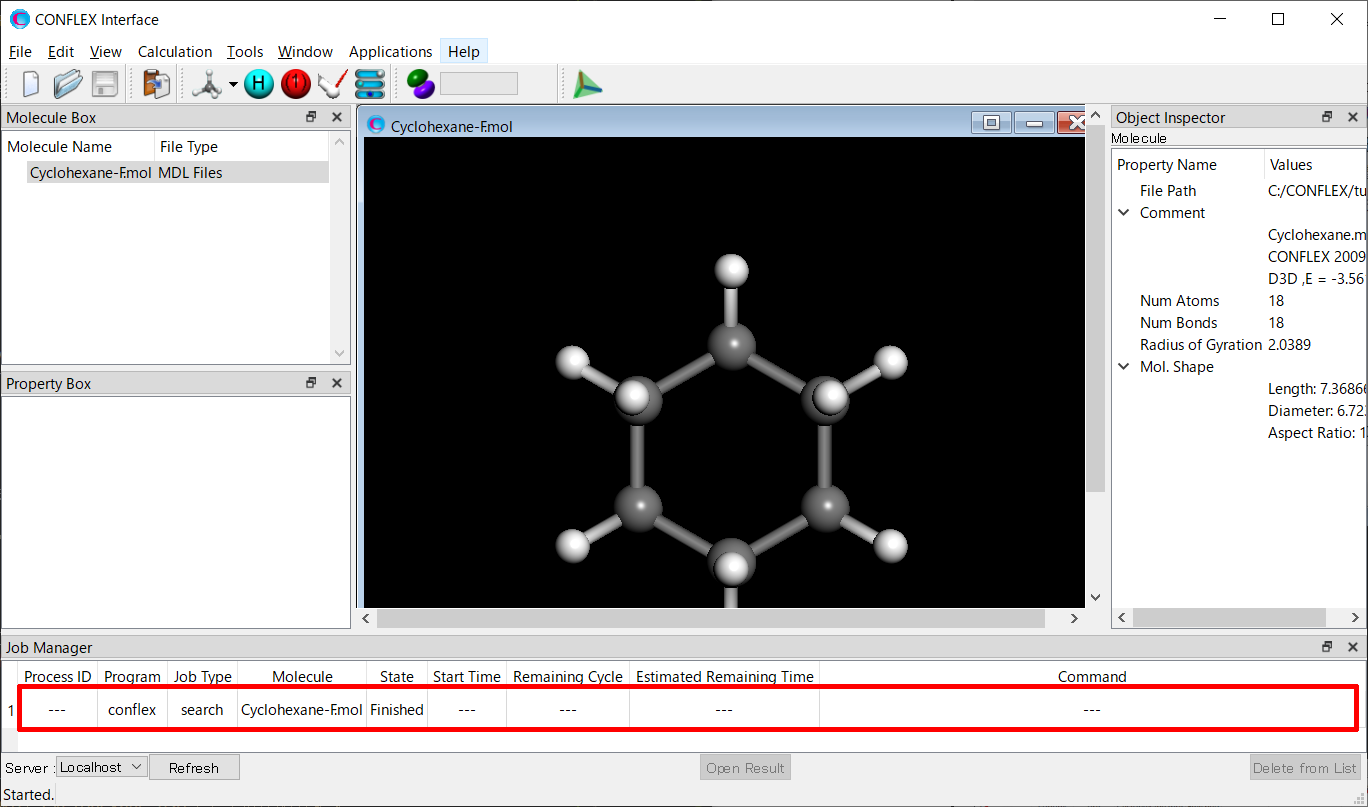
Confirm that the status of the search job is “Finished”, and then double-click the corresponding row (highlighted in red). The Cyclohexane-F.sdf file will open, displaying the conformers that were found.
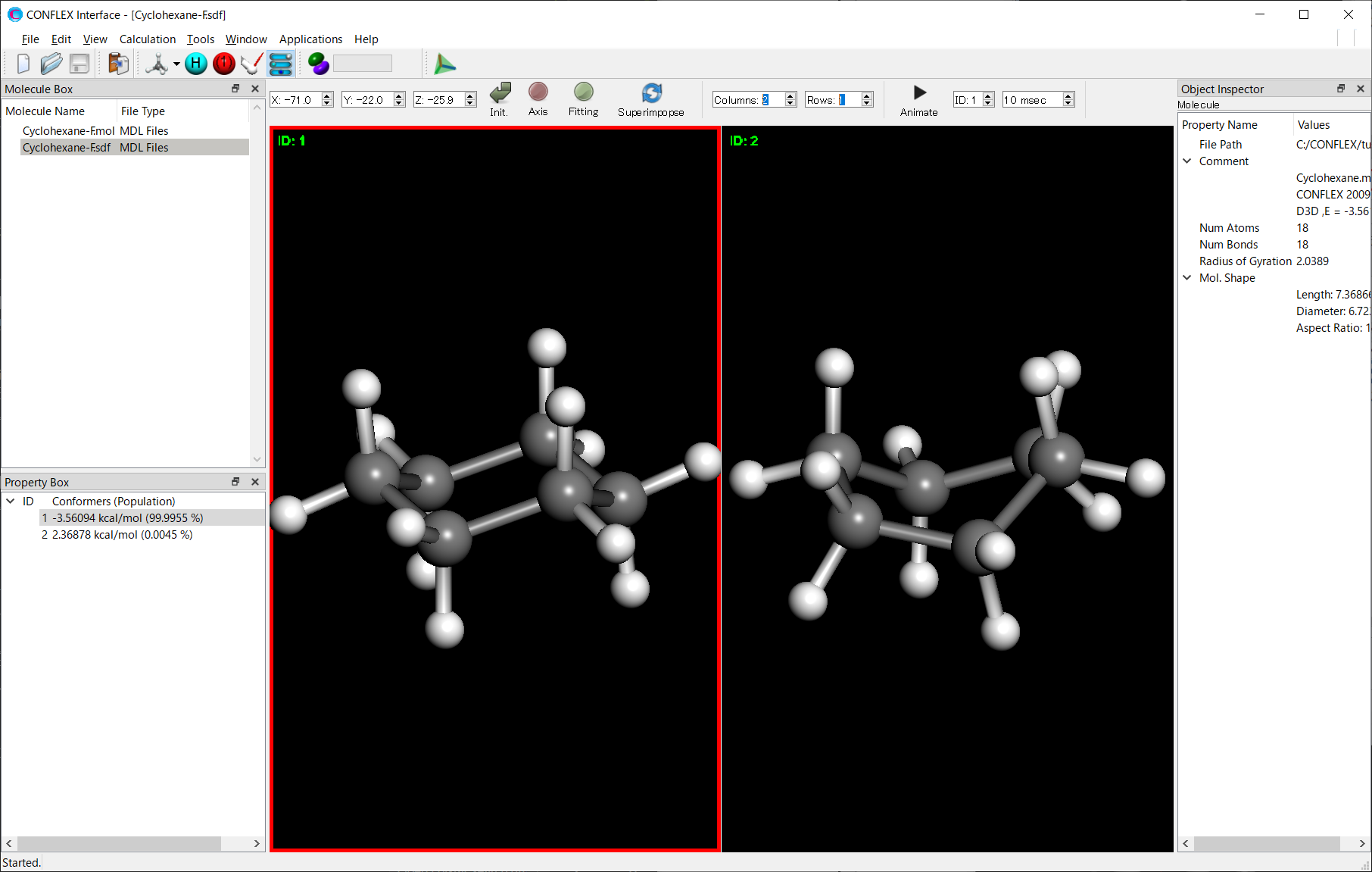
* The screen division function can be controlled by adjusting the values of [Columns/Rows] in the toolbar.
[If you executed the calculation using command line]
The Cyclohexane-F.sdf file is located in the folder containing the input files. Open the Cyclohexane-F.sdf file to visualize the conformers that were found.
* The screen division function can be controlled by adjusting the values of [Columns/Rows] in the toolbar.
[Conformation search with solvent effect]
This section explains a conformational search considering the solvent effect using the GB/SA model. About explanation of GB/SA model, please refer to [Calculation with solvent effect].
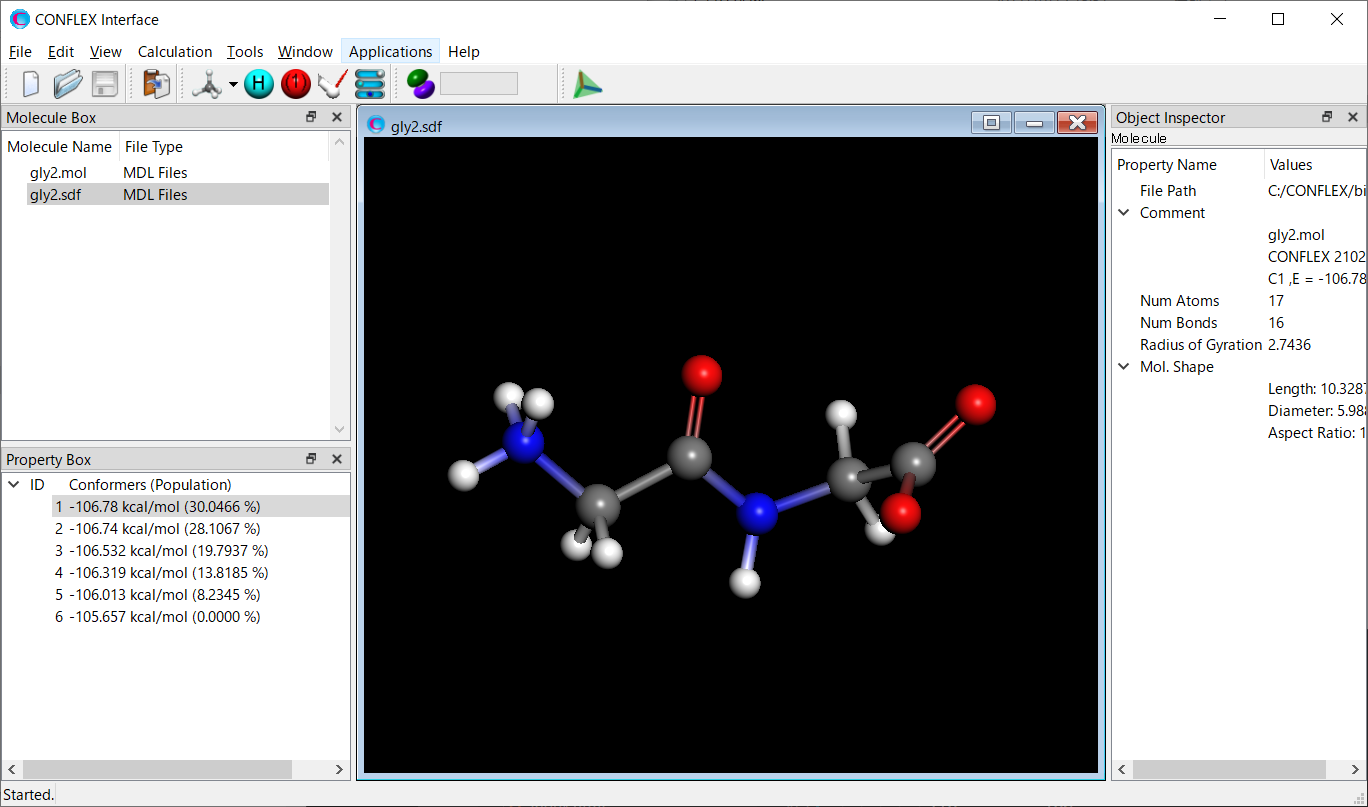
We use a zwitterionic glycine dimer.
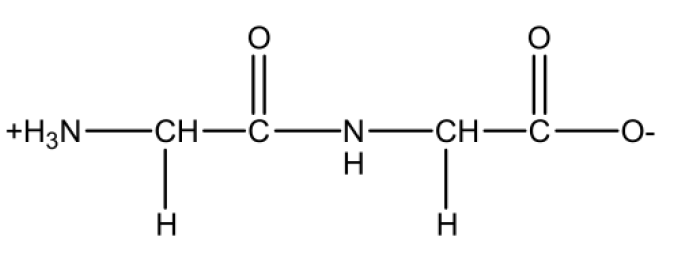
Structure data of the zwitterionic glycine dimer (gly2.mol)
gly2.mol
17 16 0 0 0 0 0 0 0 0999 V2000
-1.2219 0.9765 -2.1504 N 0 3 0 0 0 0 0 0 0 0 0 0
0.2781 0.9765 -2.1504 C 0 0 0 0 0 0 0 0 0 0 0 0
0.6496 2.0256 -2.1504 H 0 0 0 0 0 0 0 0 0 0 0 0
0.7803 0.2658 -0.9176 C 0 0 0 0 0 0 0 0 0 0 0 0
1.9679 0.1552 -0.7258 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.1098 -0.2537 -0.0165 N 0 0 0 0 0 0 0 0 0 0 0 0
0.3728 -0.9366 1.1681 C 0 0 0 0 0 0 0 0 0 0 0 0
0.9911 -0.2372 1.7741 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.8018 -1.4089 1.9892 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.6088 -2.0256 3.0592 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.9679 -1.1835 1.5993 O 0 5 0 0 0 0 0 0 0 0 0 0
-1.5708 0.4839 -3.0034 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.5698 0.4832 -1.2973 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.5701 1.9618 -2.1504 H 0 0 0 0 0 0 0 0 0 0 0 0
0.6489 0.4518 -3.0592 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.1047 -0.1611 -0.1772 H 0 0 0 0 0 0 0 0 0 0 0 0
0.9910 -1.8114 0.8658 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
1 12 1 0
1 13 1 0
1 14 1 0
2 3 1 0
2 4 1 0
2 15 1 0
4 5 2 0
4 6 1 0
6 7 1 0
6 16 1 0
7 8 1 0
7 9 1 0
7 17 1 0
9 10 2 0
9 11 1 0
M END
[Execution from Interface]
Open the gly2.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
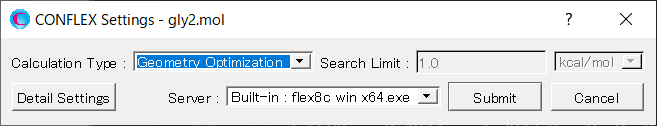
Select [Conformation Search] from the [Calculation Type:] pull-down menu in [General Settings] dialog of the detailed settings dialog.
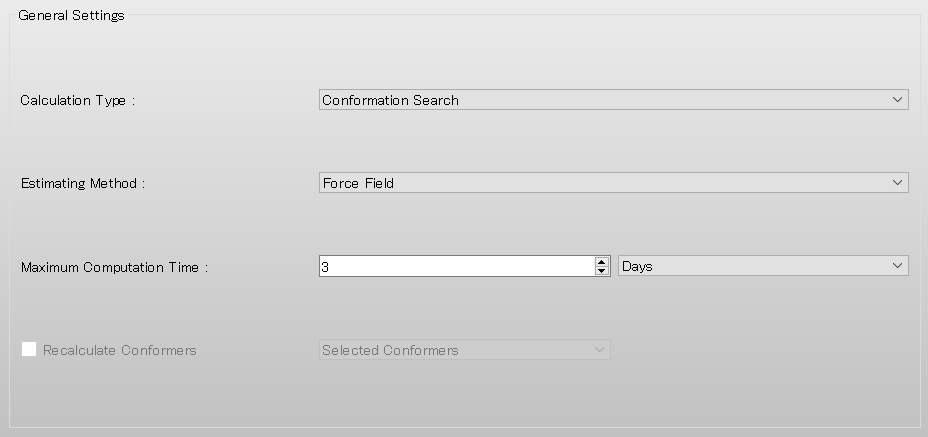
Next, select [GBSA] from the pull-down menu of [Solvent Effect] in [Force Field] dialog of the detailed settings dialog.
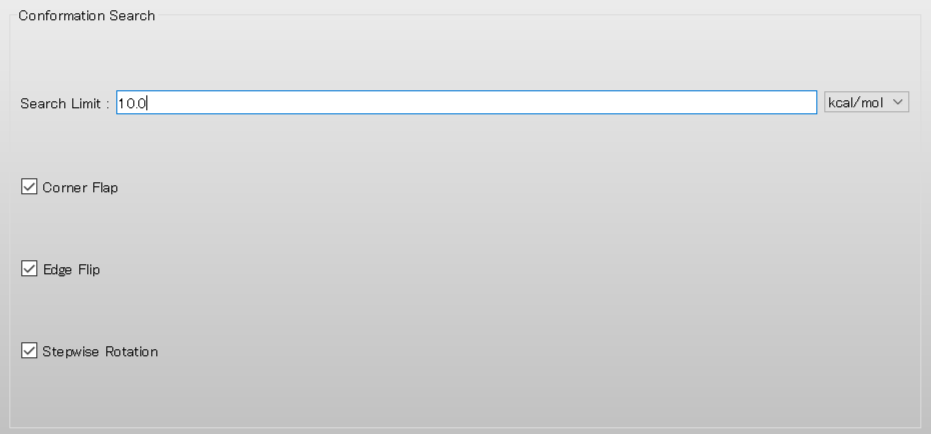
Finally, edit the value of [Search Limit:] to [10.0] in [Conformation Search] dialog. After finishing this calculation, the search of the conformational space within 10.0 kcal/mol of the most stable conformer is completed.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the gly2.ini file.
gly2.ini file
MMFF94S GBSA CONFLEX SEL=10.0
| Keyword | Explanation |
|---|---|
| MMFF94S | Use MMFF94s force filed |
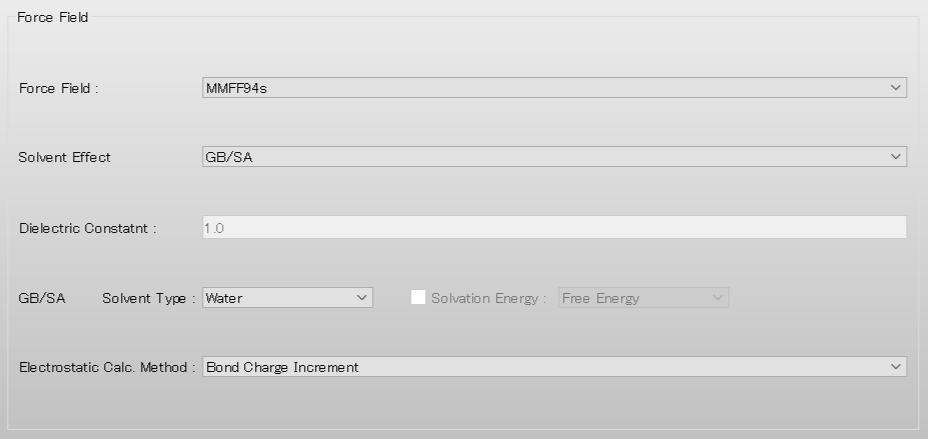
| GBSA | Introduce solvent effect by GB/SA model |
| CONFLEX | Execute a conformation search |
| SEL=10.0 | Search limit. Conformers within 10.0 kcal/mol of the most stable conformer are selected as the initial structures. |
Store the gly2.mol and gly2.ini files in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par gly2enter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
The structure of the most stable conformer is shown below. About how to visualize that, please refer to [Visualization of calculation results].