Conformer clustering
[What is a conformer clustering ?]
Classifying conformers into several meaningful groups is called conformer clustering. With the clustering, clusters can be obtained by grouping conformers with similar conformations based on structural parameters. This allows for estimating how many conformers, close to the most stable structure or a conformation in the X-ray crystal structure, exist within a specific energy range. To perform the conformer clustering, a criterion is needed to relate the conformers. The criterion used to estimate how similar a conformer is to a target conformer is called the distance between conformations. CONFLEX can perform the conformer clustering using RMSD values of dihedral angles or atomic coordinates as structural parameters. Below is an example of conformer clustering using the RMSD value of dihedral angles as the distance between conformations.
[Clustering of all conformers of n-pentane]
This section explains how to perform conformer clustering for all conformers of n-pentane. First, we conduct a conformational search for n-pentane.
n-pentane.mol
n-pentane
17 16 0 0 0 0 0 0 0 0999 V2000
-1.2316 -1.9901 -0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0874 -1.2286 -0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.1902 0.2689 -0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.1288 1.0304 -0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.8512 2.5279 -0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.0288 -3.0844 -0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.8145 -1.7213 0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.8151 -1.7211 -0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
0.6703 -1.4973 -0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
0.6709 -1.4976 0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.7731 0.5377 0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.7737 0.5379 -0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
1.7117 0.7617 -0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
1.7123 0.7614 0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
1.8151 3.0844 -0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.2683 2.7967 0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
0.2677 2.7969 -0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
1 6 1 0
1 7 1 0
1 8 1 0
2 3 1 0
2 9 1 0
2 10 1 0
3 4 1 0
3 11 1 0
3 12 1 0
4 5 1 0
4 13 1 0
4 14 1 0
5 15 1 0
5 16 1 0
5 17 1 0
M END
[Execution from Interface]
Open the n-pentane.mol file using CONFLEX Interface.
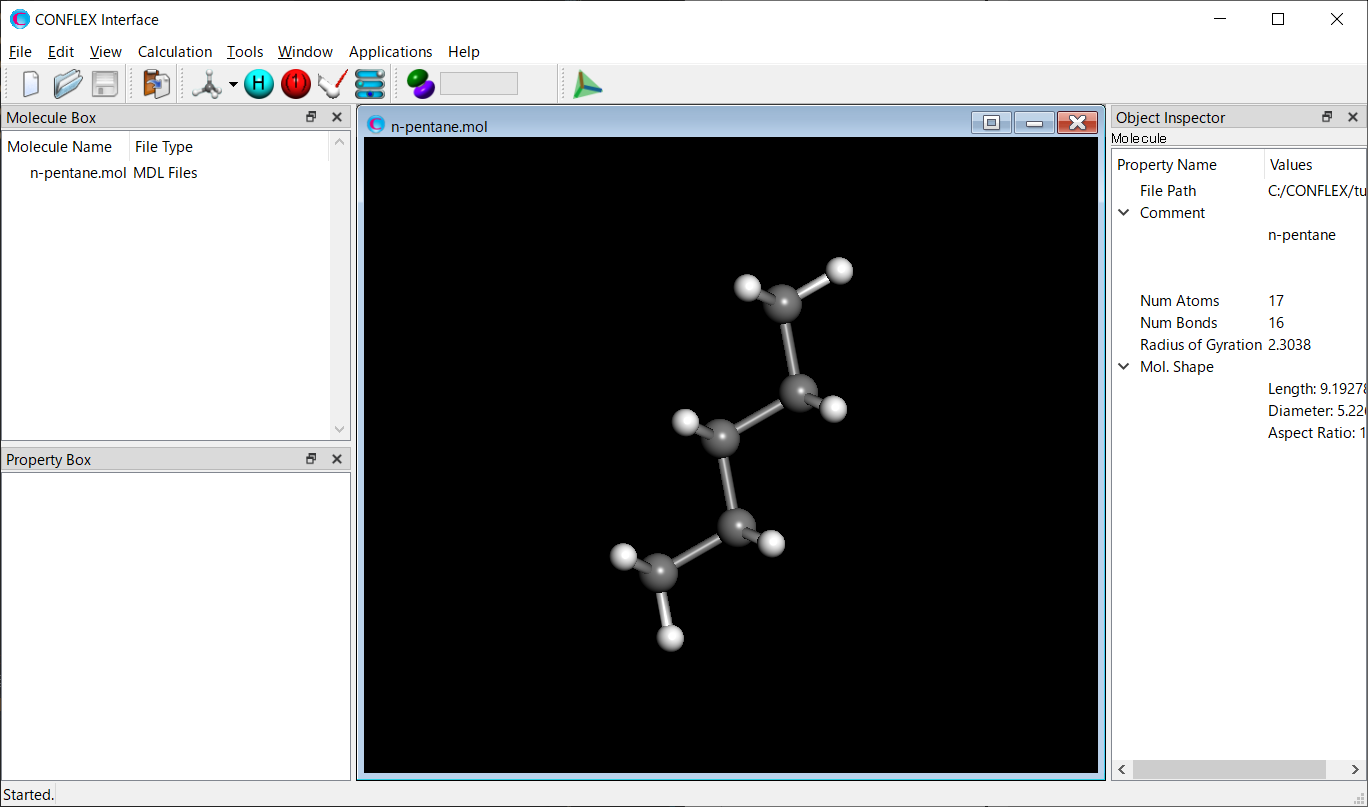
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
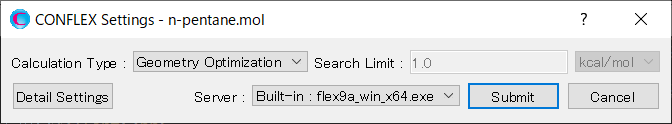
In [General Settings] dialog on the detailed settings dialog, select [Conformation Search] from the [Calculation Type:] pull-down menu.
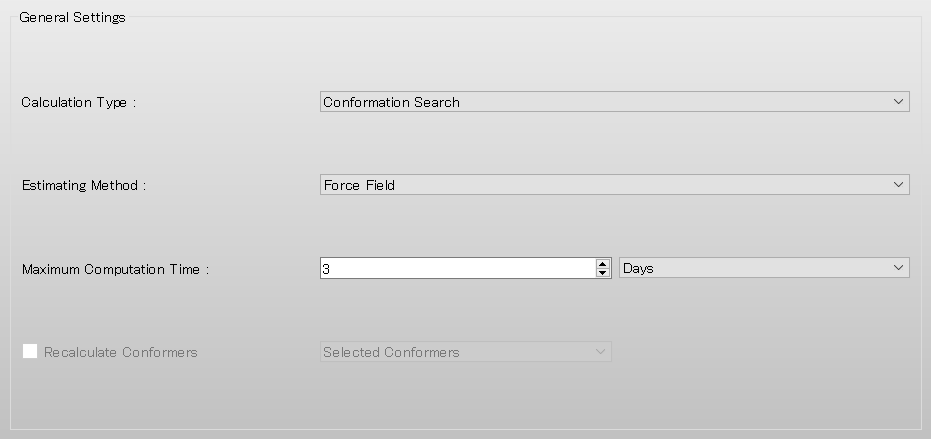
Next, edit the value of [Search Limit:] to 4.0 in the [Conformation Search] dialog. Once the calculation settings are complete, click .
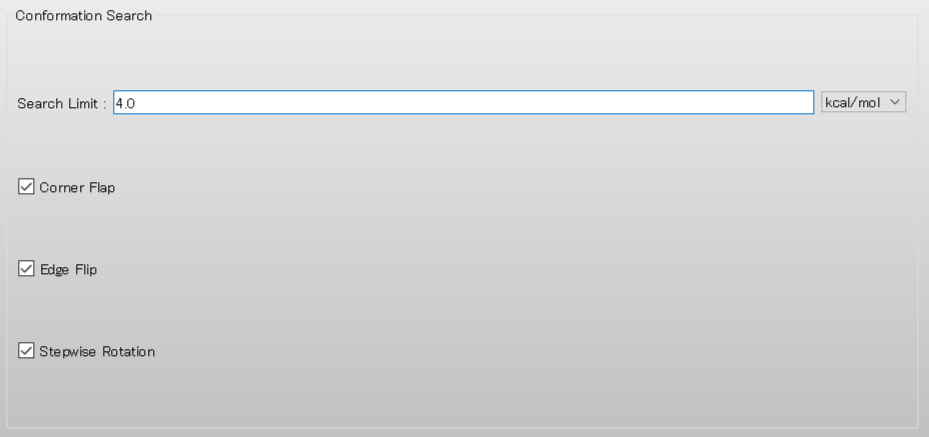
A dialog with the keywords for the calculation settings will be displayed. Add [CHECK=(TORSION,NOENERGY)] keyword to the dialog. This keyword indicates that the RMSD of dihedral angles around bonds in the backbone, rather than energy, is used as the distance between conformations.
After completing the addition, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the n-pentane.ini file.
n-pentane.ini file
MMFF94S CONFLEX SEL=4.0 CHECK=(TORSION,NOENERGY)
Explanations of each keyword are below.
| Keyword | Explanation |
|---|---|
| MMFF94S | Use MMFF94s force field |
| CONFLEX | Execute a conformation search |
| SEL=4.0 | Search limit sets to 4.0 kcal/mol. |
| CHECK=(TORSION,NOENERGY) | This keyword indicates that the RMSD of dihedral angles around bonds in the backbone, rather than energy, is used as the distance between conformations. |
Store the two files of n-pentane.mol and n-pentane.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par n-pentaneenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
After completing the search, we obtain 11 conformers. The dihedral angles of C-C-C-C for each conformer are listed below.
Table: Dihedral angles of C-C-C-C of each conformer
| No. | Steric E | Dihedral angle | |
|---|---|---|---|
| 1-2-3-4 | 2-3-4-5 | ||
| 1 | -5.2718 | -180.00 | 180.00 |
| 2 | -4.4419 | 175.69 | 65.65 |
| 3 | -4.4419 | -65.65 | -175.69 |
| 4 | -4.4419 | 65.65 | 175.69 |
| 5 | -4.4419 | -175.69 | -65.65 |
| 6 | -3.8487 | 60.27 | 60.27 |
| 7 | -3.8487 | -60.27 | -60.27 |
| 8 | -1.5718 | -64.48 | 95.34 |
| 9 | -1.5718 | 95.34 | -64.48 |
| 10 | -1.5718 | -95.34 | 64.48 |
| 11 | -1.5718 | 64.48 | -95.34 |
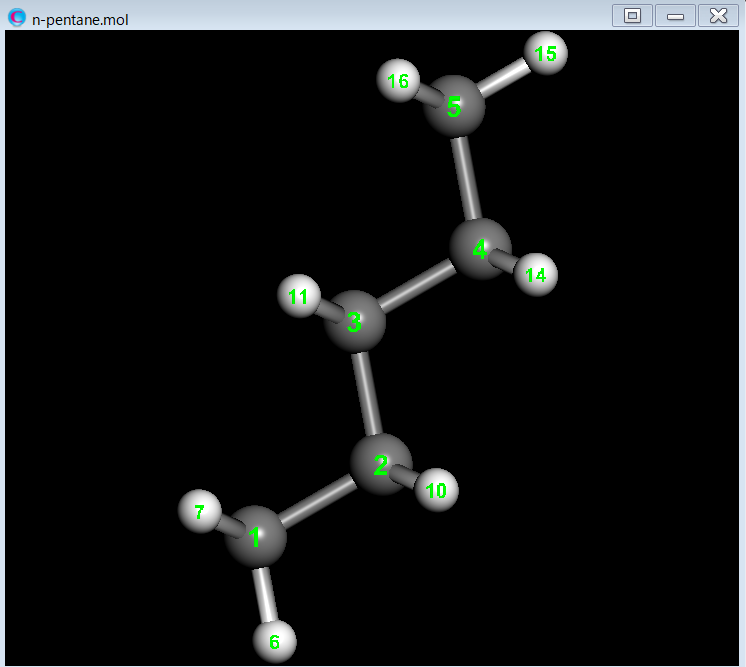
Next, we perform conformer clustering of the 11 conformers using the dihedral angle around the C2-C3 bond as the distance between conformations.
[Execution from Interface]
Open the n-pentane.mol file using CONFLEX Interface, select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
After that, click in the detailed settings dialog.
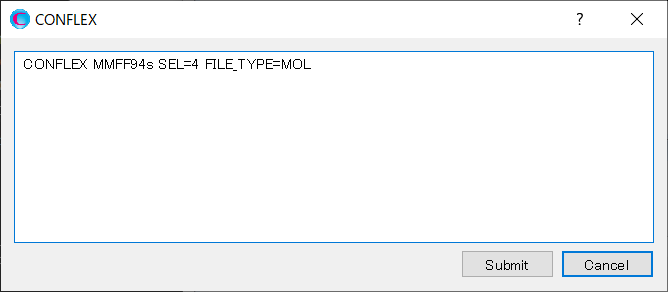
Edit the contents of the dialog as shown below, and then click to start the calculation.
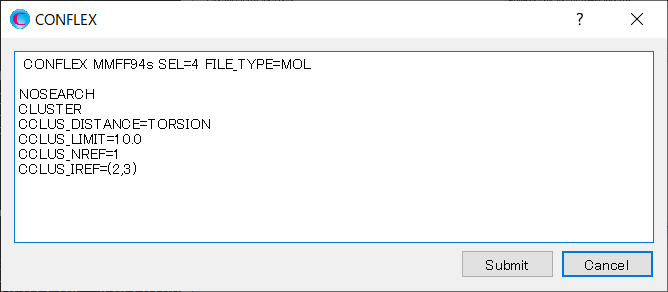
Explanations of each keyword are below.
| Keyword | Explanation |
|---|---|
| NOSEARCH | Do not perform a conformation search |
| CLUSTER | Perform a conformer clustering |
| CCLUS_DISTANCE=TORSION | Use dihedral angle as the distance between conformations |
| CCLUS_LIMIT=10.0 | Group conformers with the distance between conformations within 10.0. |
| CCLUS_NREF=1 | The number of bonds to use as a criterion for clustering. |
| CLUS_IREF=(2,3) | Serial number of atoms consisting bond to use as a criterion for clustering. |
[Execution from command line]
Edit the contents in the n-pentane.ini file as shown below.
n-pentane.ini file
MMFF94S CONFLEX SEL=4.0 CHECK=(TORSION,NOENERGY) NOSEARCH CLUSTER CCLUS_DISTANCE=TORSION CCLUS_LIMIT=10.0 CCLUS_NREF=1 CCLUS_IREF=(2,3)
Explanations of each keyword are below.
| Keyword | Explanation |
|---|---|
| NOSEARCH | Do not perform conformation search |
| CLUSTER | Perform conformer clustering |
| CCLUS_DISTANCE=TORSION | Use dihedral angle as the distance between conformations |
| CCLUS_LIMIT=10.0 | Group conformers with the distance between conformations within 10.0. |
| CCLUS_NREF=1 | The number of bonds to use as a criterion for clustering. |
| CLUS_IREF=(2,3) | Serial number of atoms consisting bond to be used as a criterion for clustering. |
Store the three files of n-pentane.mol, n-pentane.ini, and n-pentane.fxf in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par n-pentaneenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
After compliting the calculation, the results of the conformer clustering are output in the n-pentane.clu file. The fist part in this file shows the number of conformers and the index of the dihedral angle using as the distance between conformations.
=-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-=
CONFLEX CONFORMATIONAL CLUSTERING FILE
=-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-==-=
==============================================================================
# CLUSTERING INFORMATION
==============================================================================
CLUSTERING METHOD: SINGLE LINKAGE
NUMBER OF CONFORMERS CLUSTERED = 11 CONFORMERS (TOTAL 11 CONFORMERS)
DISTANCE (SIMILARITY) INDEX: TORSIONAL DISTANCE
DISTANCE DEFINITIONS: 1 TORSIONS
1: 1- 2- 3- 4
==============================================================================
Next, the distances between each pair of conformations are listed. The [SORTED NUMBER] corresponds to the order of energy, and the [CID NUMBER] corresponds to the order in which the conformers were found during the conformation search. From this list, we can see that the distance between the 4th and 9th conformers is 1.1717, based on the C1-C2-C3-C4 dihedral angle.
==============================================================================
# DISTANCE MATRIX ELEMENTS
==============================================================================
NUMBER OF DISTANCE MATIRX ELEMENTS = 55
SORTED NUMBER CID NUMBER DISTANCE
------------------ ------------------ ----------------------------
I J I J RMSD MAXD DRMSD
4 9 3 9 1.1717 -1.1717 0.0000
3 8 2 6 1.1717 1.1717 0.0000
6 9 7 9 4.2046 4.2046 3.0329
7 8 8 6 4.2046 -4.2046 0.0000
1 2 1 5 4.3141 -4.3141 0.1095
Finally, the results of conformer clustering with CCLUS_LIMIT=10.0 are output. The numbers in this table represent [CID NUMBER], and they are listed in order of energy.
==============================================================================
# RESULT - 1 IN CID NUMBER
# MIN= 1, MAX= 3, AVERAGE= 2.00, DISPERSION= 5.640
==============================================================================
DISTANCE THRESHOLD= 10.00
NCLUSTERS= 5
SIZE= 3
1 5 4
SIZE= 3
2 8 6
SIZE= 3
3 7 9
SIZE= 1
11
SIZE= 1
10
==============================================================================
Next, using the results of the conformational search obtained above, we perform conformer clustering of the 11 conformers using two dihedral angles as the distance between conformations.
[Execution from Interface]
With the n-pentane.mol file open in CONFLEX Interface, select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
After that, click in the detailed settings dialog.
Edit the contents of the dialog as shown below, and then click to start the calculation.
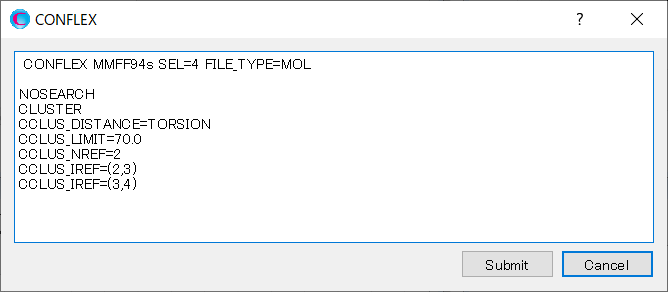
Here, CCLUS_LIMIT=70.0 is set, and the conformers are grouped with the distance between conformations within 70.0. [CCLUS_NREF=2] is set, and [CCLUS_IREF=(3,4)] is added. This means that the dihedral angle around the C3-C4 bond is included as the criterion for clustering.
[Execution from command line]
Edit the contents in the n-pentane.ini file as shown below.
n-pentane.ini file
MMFF94S CONFLEX SEL=4.0 CHECK=(TORSION,NOENERGY) NOSEARCH CLUSTER CCLUS_DISTANCE=TORSION CCLUS_LIMIT=70.0 CCLUS_NREF=2 CCLUS_IREF=(2,3) CCLUS_IREF=(3,4)
Here, CCLUS_LIMIT=70.0 is set, and the conformers are grouped with the distance between conformations within 70.0. [CCLUS_NREF=2] is set, and [CCLUS_IREF=(3,4)] is added. This means that the dihedral angle around the C3-C4 bond is included as the criterion for clustering.
Store the three files of n-pentane.mol, n-pentane.ini, and n-pentane.fxf in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par n-pentaneenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
Depending on the setting changes, the distance between conformations will change as shown below.
==============================================================================
# CLUSTERING INFORMATION
==============================================================================
CLUSTERING METHOD: SINGLE LINKAGE
NUMBER OF CONFORMERS CLUSTERED = 11 CONFORMERS (TOTAL 11 CONFORMERS)
DISTANCE (SIMILARITY) INDEX: TORSIONAL DISTANCE
DISTANCE DEFINITIONS: 2 TORSIONS
1: 1- 2- 3- 4
2: 2- 3- 4- 5
==============================================================================
# DISTANCE MATRIX ELEMENTS
==============================================================================
NUMBER OF DISTANCE MATIRX ELEMENTS = 55
SORTED NUMBER CID NUMBER DISTANCE
------------------ ------------------ ----------------------------
I J I J RMSD MAXD DRMSD
9 11 9 11 30.8622 30.8622 0.0000
8 10 6 10 30.8622 -30.8622 0.0000
4 9 3 9 62.9213 88.9765 32.0591
3 8 2 6 62.9213 -88.9765 0.0000
2 10 5 10 62.9213 88.9765 0.0000
5 11 4 11 62.9213 -88.9765 0.0000
Since CCLUS_LIMIT=70.0 was set, 5, 10, 2, and 6 and 3, 9, 4, and 11 in CID NUMBER belong to same group, respectively.
==============================================================================
# RESULT - 1 IN CID NUMBER
# MIN= 1, MAX= 4, AVERAGE= 2.00, DISPERSION= 6.840
==============================================================================
DISTANCE THRESHOLD= 70.00
NCLUSTERS= 5
SIZE= 1
1
SIZE= 4
5 10 2 6
SIZE= 4
3 9 4 11
SIZE= 1
7
SIZE= 1
8
==============================================================================
[Clustering of all conformers of β-Glucose]
Clustering of all conformers for β-Glucose is performed using the dihedral angles of the 6-membered ring as the distance between conformations.
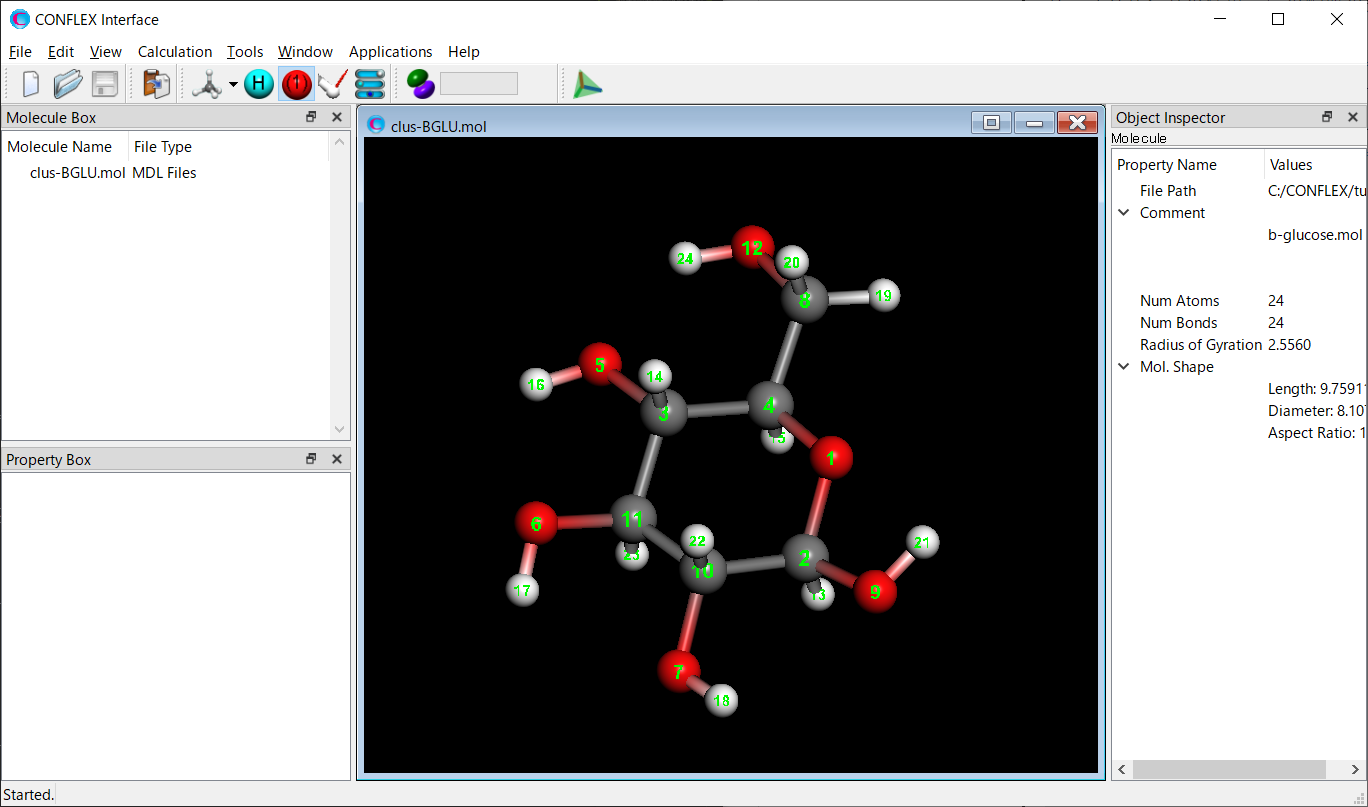
Store three files of clus-BGLU.mol, clus-BGLU.ini, and clus-BGLU.fxf in a single folder.
These files are located in the Sample_Files folder in the folder where CONFLEX is installed (Sample_Files\CONFLEX\clustering\b-glucose).
[Execution from Interface]
Open the clus-BGLU.mol file using CONFLEX Interface.

Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears.
After that, click in the detailed settings dialog. A dialog containing the keywords for the calculation settings will be displayed.
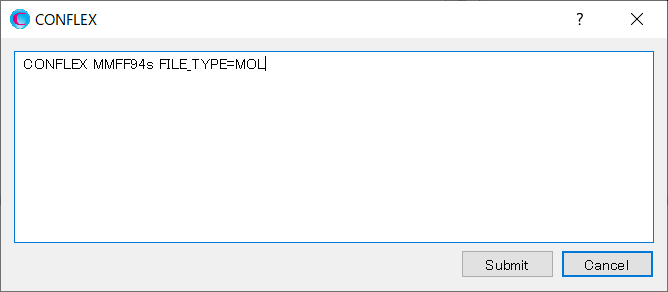
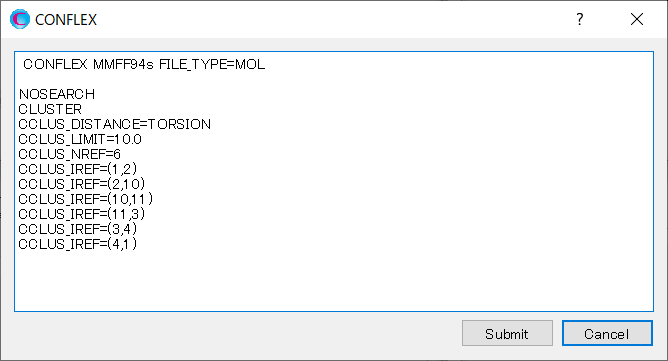
Add keywords to the dialog as shown below, and then click to start the calculation.
[Execution from command line]
The calculation settings have already written in the clus-BGLU.ini file.
clus-BGLU.ini file
MMFF94S CONFLEX NOSEARCH CLUSTER CCLUS_DISTANCE=TORSION CCLUS_LIMIT=10.0 CCLUS_NREF=6 CCLUS_IREF=(1,2) CCLUS_IREF=(2,10) CCLUS_IREF=(10,11) CCLUS_IREF=(11,3) CCLUS_IREF=(3,4) CCLUS_IREF=(4,1)
Execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par clus-BGLUenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
The conformers are classified into 7 groups based on the conformation of 6-members ring.
==============================================================================
# RESULT - 1 IN CID NUMBER
# MIN= 1, MAX= 122, AVERAGE= 31.00, DISPERSION= 2499.816
==============================================================================
DISTANCE THRESHOLD= 10.00
NCLUSTERS= 7
SIZE= 122
22 3 1 28 21 4
12 13 2 59 14 127
40 35 64 131 20 68
57 33 74 58 82 88
135 75 54 106 56 34
92 5 55 65 97 139
67 144 90 102 116 133
39 101 72 132 111 83
79 94 134 161 45 46
103 86 151 100 167 80
118 108 121 145 73 155
149 173 126 95 140 49
113 87 66 51 107 62
98 52 125 61 142 124
156 141 112 168 53 123
89 117 166 122 160 60
105 154 93 148 170 78
150 115 96 176 153 171
157 143 165 162 169 164
175 158 163 177 172 182
178 180
SIZE= 33
76 31 29 99 137 104
119 109 26 114 17 188
9 23 38 192 6 77
146 37 91 110 210 48
44 174 43 15 69 42
181 203 47
SIZE= 31
50 16 30 130 138 85
71 84 147 159 129 41
152 136 179 120 36 186
70 25 128 195 81 185
184 201 63 191 8 194
193
SIZE= 4
32 24 10 7
SIZE= 26
205 214 190 197 208 207
218 189 202 212 187 198
183 199 216 211 209 220
217 204 215 196 213 206
200 219
SIZE= 3
27 19 11
SIZE= 1
18
==============================================================================
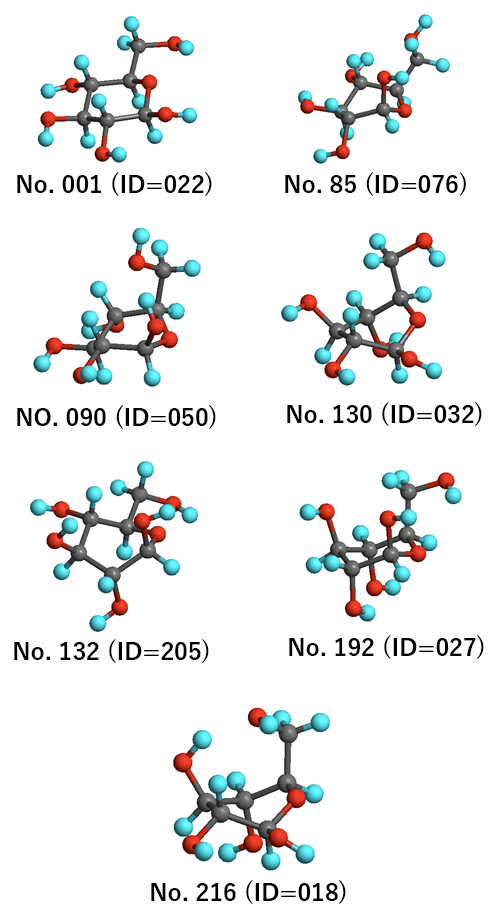
The most stable structures in each group are shown below.