Crystal surface analysis
[Calculation overview]
Analysis of a crystal surface is essential for understanding phenomena in solids, such as dissolution, sublimation, growth, and more. CONFLEX can calculate the specific surface energy of a crystal plane, as well as the sum of interatomic interaction energies with respect to a molecule in or on a crystal plane.
- Calculation of specific surface energy
- Calculation of interaction energy of molecule on/in crystal plane
[Calculation of specific surface energy]
CONFLEX calculates the specific surface energy of a crystal plane (Ehkl) using the following equation [Kim, Y., Matsumoto, M., Machida, K., Chem. Pharm. Bull. 33(10), 4125 (1985).].
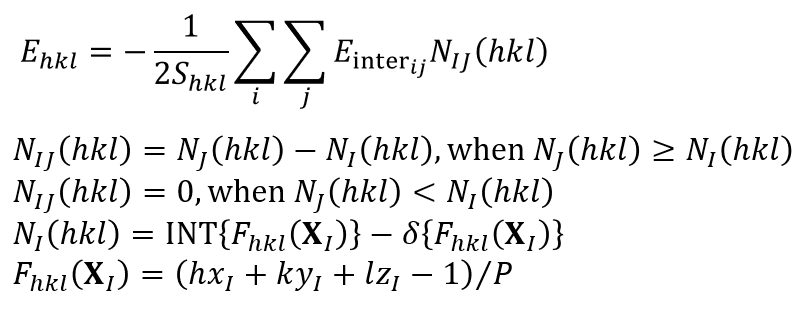
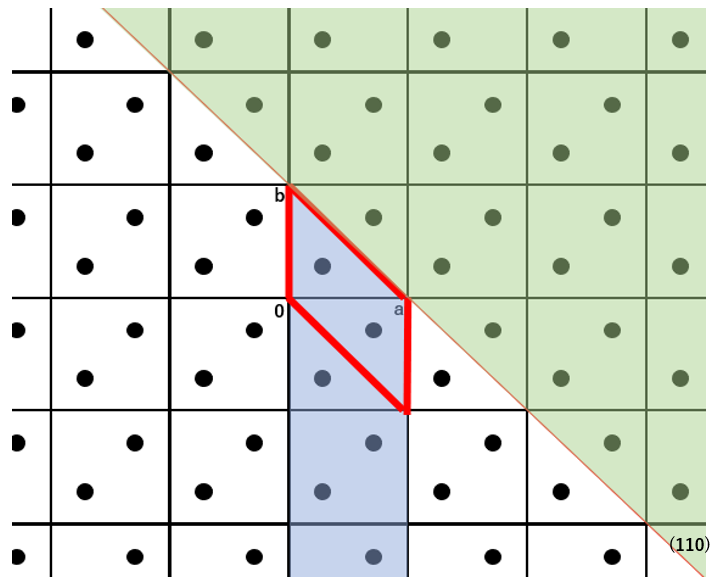
In the equation, the Shkl refers to the exposed area of the basic unit cell, the Einterij is the interatomic interaction energy between atom i and atom j. The i runs over all atoms in the basic unit cell, and the j runs over all atoms of molecules within cutoff distance Rcrystal from the molecule containing the atom i. The I denotes the molecular index of the molecule containing the atom i, while the J indicates the molecular index of the molecule containing the atom j. The NIJ(hkl) is the number of equivalent I-J molecular pairs that exist across the specified crystal plane. The XI represents the coordinates of the center of molecule I in the fractional coordinate system. The P is greatest common divisor of the absolute values of h, k, and l. The INT(F) refers to the integer part of real number F, and the δ(F) is 0 when the F is positive or 1 when the F is negative. In other words, the double summation in the equation represents the sum of the intermolecular interaction energies between all molecules within one unit cell on one side of the specified crystal plane, including the columnar region behind it (blue area), and all molecules on the opposite side of the specified crystal plane (green area). This corresponds to the surface energy per unit cell. The specific surface energy is then calculated by dividing the surface energy per unit cell by the exposed surface area. Note that since the specific surface energy is calculated based on the surface energy per unit cell, the calculation can only be performed using a model with space group P1.
[How to perform the calculation]
We use a crystal of hydroxy malonic acid, one of the malonic acid derivatives [Roelofsen, G.; Kanters, J.A.; Kroon, J.; Doesburg, H.M.; Koops, T. Acta Cryst.1978, B34, 2565.].
The input file (tartronicacid.cmf) is shown below. You can find this file in the Sample_Files folder located in the CONFLEX installation directory (Sample_Files\CONFLEX\crystal\plane\in_plane\tartronicacid.cmf).
tartronicacid.cmf file
data_Tartronicacid _symmetry_cell_setting ORTHORHOMBIC _symmetry_space_group_name_H-M 'P212121 ' _ccdc_symmetry_space_group_name P212121 _symmetry_Int_Tables_number 19 loop_ _symmetry_equiv_pos_site_id _symmetry_equiv_pos_as_xyz 1 x,y,z 2 1/2+x,1/2-y,-z 3 -x,1/2+y,1/2-z 4 1/2-x,-y,1/2+z _cell_length_a 4.49400 _cell_length_b 8.81900 _cell_length_c 10.88200 _cell_angle_alpha 90.00000 _cell_angle_beta 90.00000 _cell_angle_gamma 90.00000 _cell_formula_units_Z 4 _cell_volume 431.28180 _exptl_crystal_density_diffrn 1.84821 loop_ _ccdc_atom_site_atom_id_number _atom_site_label _atom_site_type_symbol _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z _ccdc_atom_site_symmetry _ccdc_atom_site_base 1 O1 O 1.12990 -0.13910 0.36040 1_555 1 2 O2 O 0.97510 0.09280 0.30700 1_555 2 3 O3 O 1.01480 0.11550 0.66290 1_555 3 4 O4 O 1.13030 -0.12820 0.62810 1_555 4 5 O5 O 0.57240 0.09790 0.48970 1_555 5 6 C1 C 0.97750 -0.01540 0.37600 1_555 6 7 C2 C 0.78810 -0.01520 0.49230 1_555 7 8 C3 C 0.99010 -0.00160 0.60420 1_555 8 9 H1 H 0.66800 -0.11200 0.49600 1_555 9 10 H2 H 0.60500 0.14900 0.45600 1_555 10 11 H3 H 1.23700 -0.13800 0.31000 1_555 11 12 H4 H 1.27100 -0.12000 0.68000 1_555 12 loop_ _atom_id _atom_type _atom_attach_nh _atom_attach_h _atom_charge 1 O 1 1 0 2 O 1 0 0 3 O 1 0 0 4 O 1 1 0 5 O 1 1 0 6 C 3 0 0 7 C 3 1 0 8 C 3 0 0 loop_ _bond_id_1 _bond_id_2 _bond_type_ccdc _bond_environment 1 6 S chain 1 11 S chain 2 6 D chain 3 8 D chain 4 8 S chain 4 12 S chain 5 7 S chain 5 10 S chain 6 7 S chain 7 8 S chain 7 9 S chain
[Execution from Interface]
Open the tartronicacid.cmf file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
First, in [General Settings] dialog of the detailed settings dialog, select [Molecular Crystal] from the pull-down menu of [Calculation Type:].
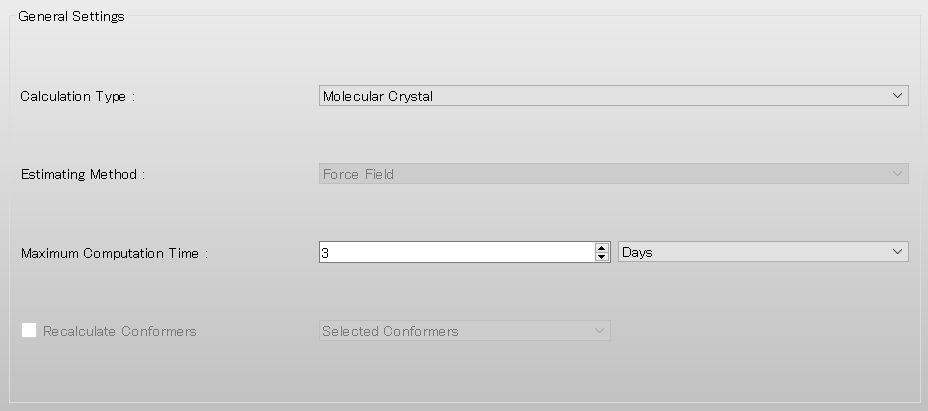
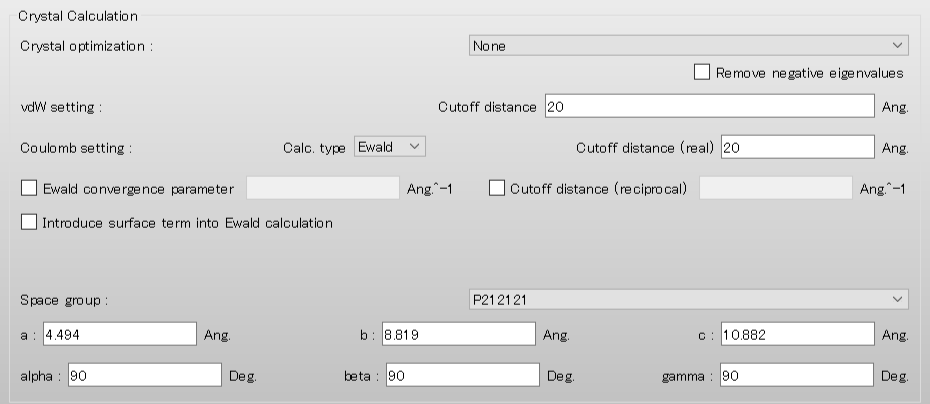
Next, select [None] from the pull-down menu of [Crystal Optimization:] in the [Crystal Calculation] dialog. If you want to optimize the crystal structure before performing the surface analysis, select [ALL] or another appropriate option.
Next, configure the settings for the surface analysis using the [Crystal Surface] tab in the [Crystal Property] dialog.
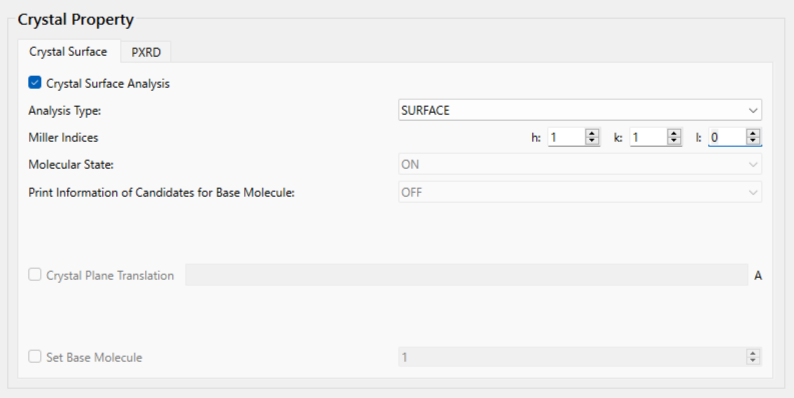
First, check the [Crystal Surface Analysis] checkbox.
Next, set the Miller indices of the crystal plane under [Miller Indices]. Here, set h = 1, k = 1, and l = 0.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the tartronicacid.ini file.
tartronicacid.ini file
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(1,1,0) MAKEP1CELL
[CRYSTAL] indicates that a crystal calculation will be executed.
[CRYSTAL_OPTIMIZATION=NONE] means that the crystal structure will not be optimized. If you want to optimize the crystal structure before the analysis, select [ALL] or another appropriate option.
[CRYSTAL_PLANE=(1,1,0)] specifies that the specific surface energy of the (110) crystal plane will be calculated.
[MAKEP1CELL] means that the initial space group will be converted to P1.
Store the two files of tartronicacid.cmf and tartronicacid.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par tartronicacidenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
[Calculation results]
The specific surface energy of the (110) crystal plane, E110, is output in the tartronicacid.bso file as shown below.
*** SPECIFIC SURFACE ENERGY CALCULATION
* CHANGE TO CUTOFF METHOD IN CALCULATION OF ELECTROSTATIC INTERACTIONS
SURFACE AREA SPECIFIC
(h k l) d_hkl (Ang.) (Ang.**2) SURFACE ENERGY (mJ/m**2)
1 1 0 4.0041 107.7103 216.2389
You can also obtain a PDB file of the model that includes one basic unit cell on one side of the specified crystal plane and a hemispherical crystal on the other side. The file is named in the format: (input file name)_hkl.pdb. By opening the tartronicacid_110.pdb file in the CONFLEX Interface, you can see the model.
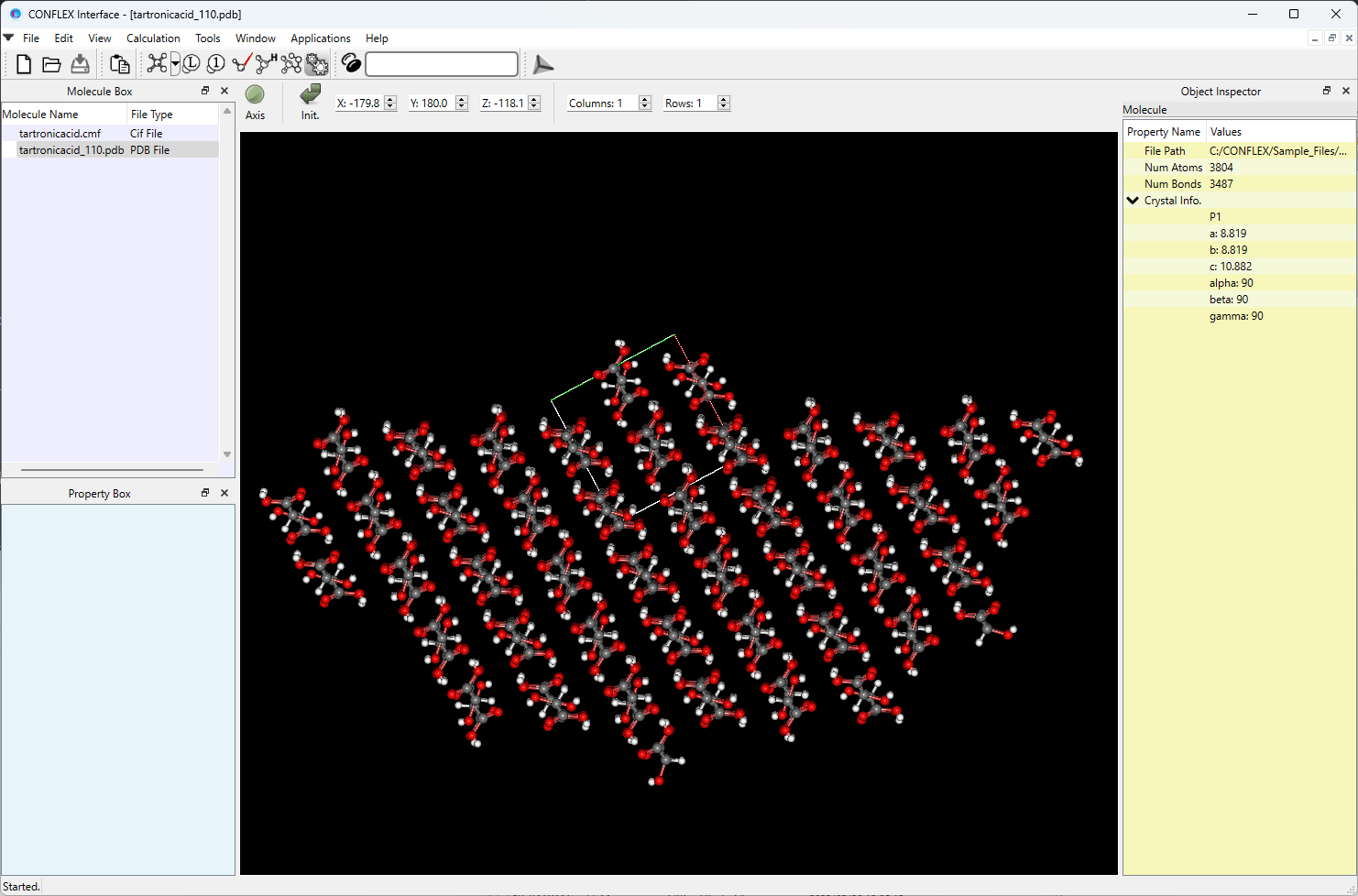
[Calculation of interaction energy of molecule on/in crystal plane]
For example, if you specify the (100) plane, CONFLEX constructs a hemispherical crystal with the (100) plane exposed (as shown in the figure below) and calculates the sum of interatomic interaction energies with respect to the molecule in or on the (100) plane. The radius of the hemispherical crystal is determined by the cutoff value.
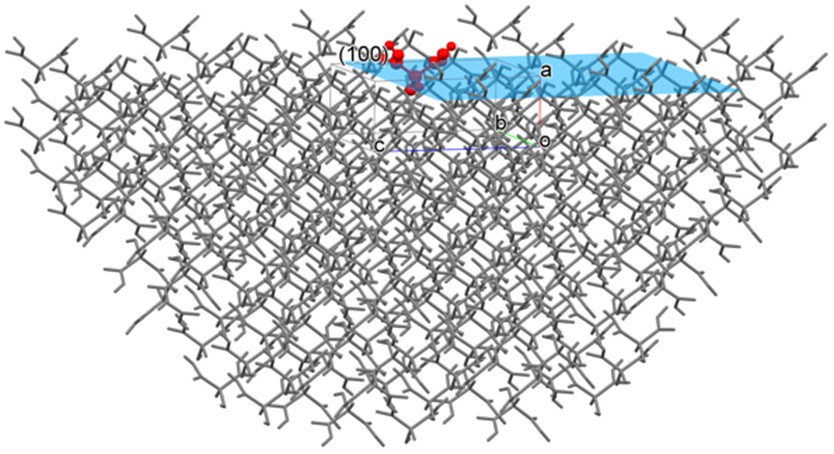

Here, Emol is the sum of the interatomic interaction energies with respect to the molecule (target molecule, the molecule shown in red in the above figure) located in or on the center of the (100) plane, N is the total number of atoms of the target molecule, and M is the total number of atoms in the hemispherical crystal excluding the target molecule.
[How to perform the calculation]
We use a crystal of hydroxy malonic acid, one of the malonic acid derivatives [Roelofsen, G.; Kanters, J.A.; Kroon, J.; Doesburg, H.M.; Koops, T. Acta Cryst.1978, B34, 2565.].
The input file (tartronicacid.cmf) is shown below. You can find this file in the Sample_Files folder located in the CONFLEX installation directory (Sample_Files\CONFLEX\crystal\plane\in_plane\tartronicacid.cmf).
tartronicacid.cmf file
data_Tartronicacid _symmetry_cell_setting ORTHORHOMBIC _symmetry_space_group_name_H-M 'P212121 ' _ccdc_symmetry_space_group_name P212121 _symmetry_Int_Tables_number 19 loop_ _symmetry_equiv_pos_site_id _symmetry_equiv_pos_as_xyz 1 x,y,z 2 1/2+x,1/2-y,-z 3 -x,1/2+y,1/2-z 4 1/2-x,-y,1/2+z _cell_length_a 4.49400 _cell_length_b 8.81900 _cell_length_c 10.88200 _cell_angle_alpha 90.00000 _cell_angle_beta 90.00000 _cell_angle_gamma 90.00000 _cell_formula_units_Z 4 _cell_volume 431.28180 _exptl_crystal_density_diffrn 1.84821 loop_ _ccdc_atom_site_atom_id_number _atom_site_label _atom_site_type_symbol _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z _ccdc_atom_site_symmetry _ccdc_atom_site_base 1 O1 O 1.12990 -0.13910 0.36040 1_555 1 2 O2 O 0.97510 0.09280 0.30700 1_555 2 3 O3 O 1.01480 0.11550 0.66290 1_555 3 4 O4 O 1.13030 -0.12820 0.62810 1_555 4 5 O5 O 0.57240 0.09790 0.48970 1_555 5 6 C1 C 0.97750 -0.01540 0.37600 1_555 6 7 C2 C 0.78810 -0.01520 0.49230 1_555 7 8 C3 C 0.99010 -0.00160 0.60420 1_555 8 9 H1 H 0.66800 -0.11200 0.49600 1_555 9 10 H2 H 0.60500 0.14900 0.45600 1_555 10 11 H3 H 1.23700 -0.13800 0.31000 1_555 11 12 H4 H 1.27100 -0.12000 0.68000 1_555 12 loop_ _atom_id _atom_type _atom_attach_nh _atom_attach_h _atom_charge 1 O 1 1 0 2 O 1 0 0 3 O 1 0 0 4 O 1 1 0 5 O 1 1 0 6 C 3 0 0 7 C 3 1 0 8 C 3 0 0 loop_ _bond_id_1 _bond_id_2 _bond_type_ccdc _bond_environment 1 6 S chain 1 11 S chain 2 6 D chain 3 8 D chain 4 8 S chain 4 12 S chain 5 7 S chain 5 10 S chain 6 7 S chain 7 8 S chain 7 9 S chain
[Execution from Interface]
Open the tartronicacid.cmf file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. A detailed settings dialog will be displayed.
First, in [General Settings] dialog of the detailed settings dialog, select [Molecular Crystal] from the pull-down menu of [Calculation Type:].
Next, select [None] from the pull-down menu of [Crystal Optimization:] in the [Crystal Calculation] dialog. If you want to optimize the crystal structure before performing the surface analysis, select [ALL] or another appropriate option.
Next, configure the settings for the surface analysis using the [Crystal Surface] tab in the [Crystal Property] dialog.
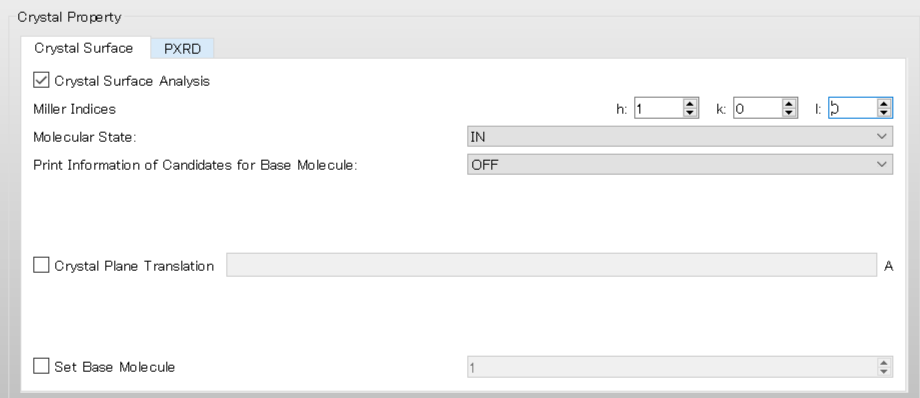
First, check the [Crystal Surface Analysis] checkbox.
Select [MOLECULE] from the pull-down menu of [Analysis Type:].
Next, set the Miller indices of the crystal plane under [Miller Indices]. Here, set h = 1, k = 0, and l = 0.
[In] under [Molecular State:] means that Emol, with respect to the molecule composing the (100) plane, is calculated.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the tartronicacid.ini file.
tartronicacid.ini file
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(1,0,0) CRYSTAL_PLANE_ANALYSIS_TYPE=MOLECULE
[CRYSTAL] indicates that a crystal calculation will be executed.
[CRYSTAL_OPTIMIZATION=NONE] means that the crystal structure will not be optimized. If you want to optimize the crystal structure before the analysis, select [ALL] or another appropriate option.
[CRYSTAL_PLANE=(1,0,0)] means the analysis is performed for the (100) plane. By default, the Emol value for the molecule composing the (100) plane is calculated.
[CRYSTAL_PLANE_ANALYSIS_TYPE=MOLECULE] means the interaction energy of the molecule in the crystal plane will be calculated.
Store the two files of tartronicacid.cmf and tartronicacid.ini in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par tartronicacidenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
[Calculation results]
After the calculation, you will find the tartronicacid_100.pdb file. This file contains the structural data of the hemispherical crystal with the (100) plane exposed. The file is named in the format: (input file name)_hkl.pdb. By opening the tartronicacid_100.pdb file in the CONFLEX Interface, you can see the hemispherical crystal.
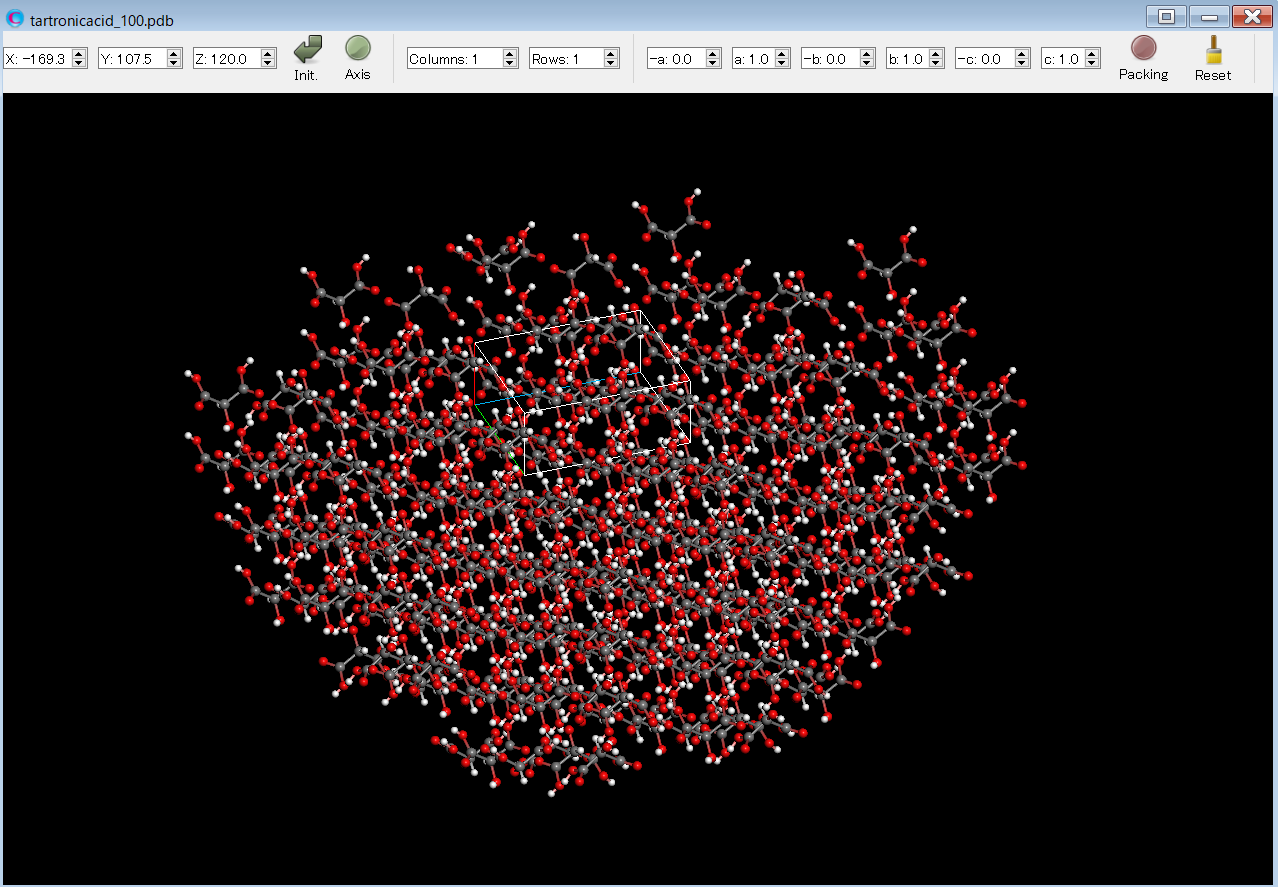
The Emol of the target molecule is shown in the tartronicacid.bso file as shown below.
* SUM OF INTERMOLECULAR INTERACTION ENERGIES OF MOLECULE
[IN] THE CRYSTAL PLANE
* CHANGE TO CUTOFF METHOD IN CALCULATION OF ELECTROSTATIC INTERACTIONS
ENERGY
H K L (KCAL/MOL)
1 0 0 -10.7323
[Regarding the alphabetical notation next to the energy]
The analysis of the (010) plane of the hydroxy malonic acid crystal is performed using the tartronicacid.cmf file. In this case, the crystal structure is not optimized.
The Emol of the target molecule is shown in the tartronicacid.bso file as shown below.
* SUM OF INTERMOLECULAR INTERACTION ENERGIES OF MOLECULE
[IN] THE CRYSTAL PLANE
* CHANGE TO CUTOFF METHOD IN CALCULATION OF ELECTROSTATIC INTERACTIONS
ENERGY
H K L (KCAL/MOL)
0 1 0 -39.2094 a
0 1 0 -38.4304 b
The target molecule is automatically determined by the program. However, when multiple unique molecules are present in the specified crystal plane, the the Emol for each unique molecule is displayed separately, with the results distinguished by alphabetical labels. For example, “a” and “b” represent results of the analysis for the (010) plane, but the target molecules used in the energy calculations are different.
[Difference of “IN” and “ON” in “Molecular State:”]
In the [Crystal Surface] tab of the [Crystal Property] dialog, the state of the target molecule differs depending on whether [Molecular State:] is set to [IN] or [ON], as shown in the following figure. When executing from the command line, this corresponds to whether [CRYSTAL_PLANE_STATE=IN] or [CRYSTAL_PLANE_STATE=ON] is written in the ini file.
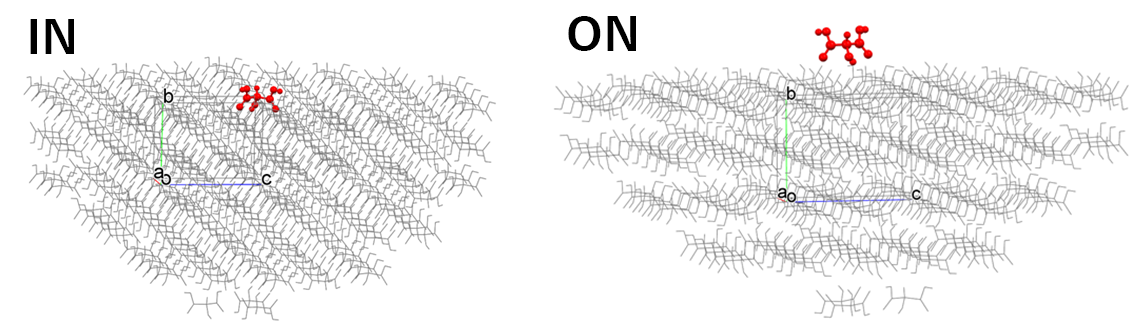
[IN] selects the molecule that composes the specified crystal plane, and the Emol is calculated for it.
[ON] selects the molecule located on the specified crystal plane, and the Emol is calculated for it.
[How to use “Crystal Plane Translation”]
The analysis of the (001) plane of the hydroxy malonic acid crystal is performed using the tartronicacid.cmf file. In this case, crystal structure optimization is not conducted. CONFLEX constructs a hemispherical crystal as shown in the figure below and calculates Emol.
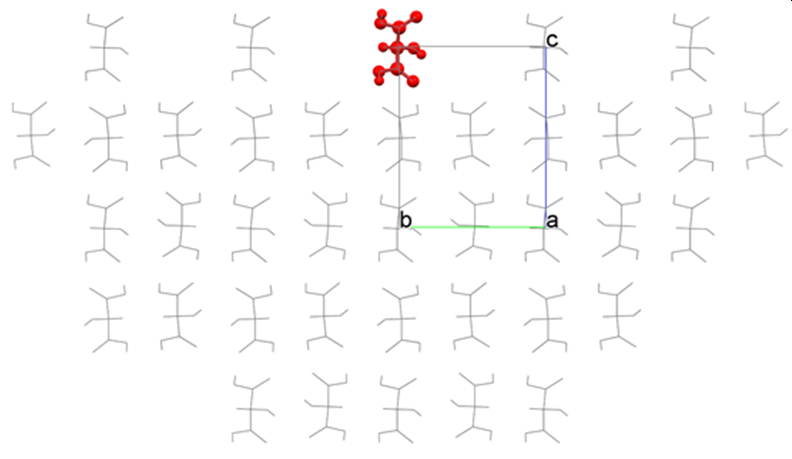
The interface of the hemispherical crystal is automatically determined by the program. However, you can change the position of the interface by setting a value for [Crystal Plane Translation] in the [Crystal Surface] tab of the [Crystal Property] dialog. In the figure above, the position of the crystal plane is “0”; the upward direction is positive, and the downward direction is negative.
For example, if you set the value of [Crystal Plane Translation] to 1.0, the interface will move in the upward direction by 1.0 Ang..
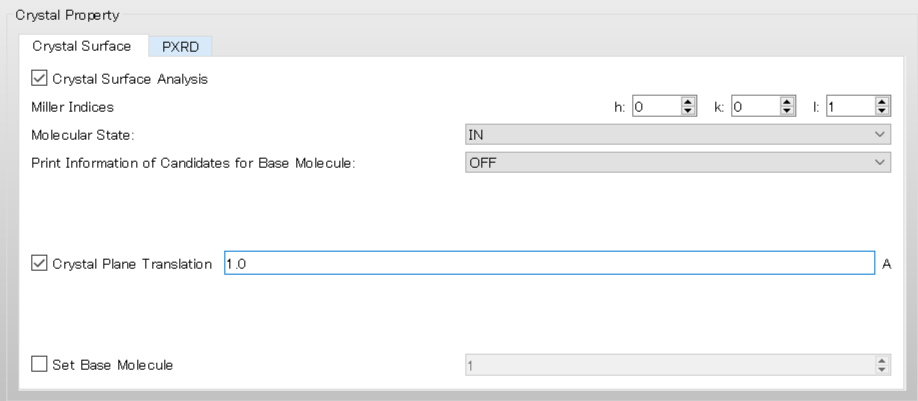
In the execution from the command line, this corresponds to including [CRYSTAL_PLANE_TRANS=1.0] in the ini file.
tartronicacid.ini file
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(0,0,1) CRYSTAL_PLANE_TRANS=1.0
As the result, a hemispherical crystal, as shown in the following figure, is constructed, and the Emol is calculated.
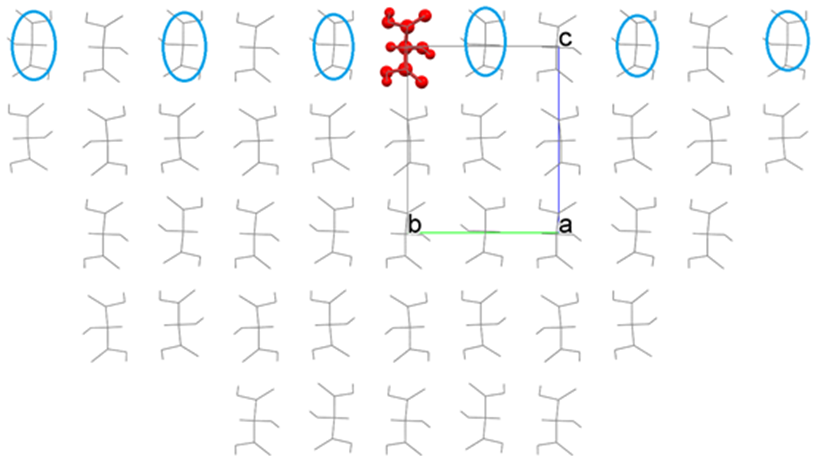
By comparing the before and after moving the interface, we can see that the molecules indicated by the blue circle are now included in the calculation of Emol due to the movement of the interface. Please adjust the position of the interface if necessary.
[How to use “Set Base Molecule”]
The target molecule is automatically determined by the program. However, you can change the target molecule by setting [Set Base Molecule] in the [Crystal Surface] tab of the [Crystal Property] dialog. In this example, an analysis of the (100) plane of the hydroxy malonic acid crystal will be explained using the tartronicacid.cmf file as the input.
First, to obtain information about the candidates for the target molecule, select [ON] from the pull-down menu of [Print Information of Candidates for Base Molecule:] in the [Crystal Surface] tab, and then perform the calculation.
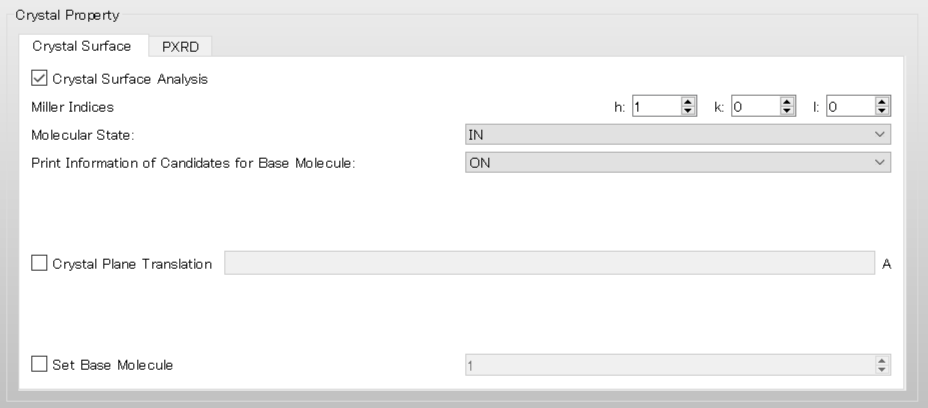
In the case of execution from the command line, add [CRYSTAL_PLANE_PRINT=ON] to the ini file and perform the calculation.
tartronicacid.ini file
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(1,0,0) CRYSTAL_PLANE_PRINT=ON
In the tartronicacid.bso file, the molecular index (IDX) and the position of the center of mass (POSITION) for the candidates are provided. In the POSITION data, the crystal plane position is “0”, with the positive value being upwards and the negative value being downwards (as shown in the following figure).
IDX POSITION (ANG.) 1 0.224367 2 0.224367 3 -0.224367 4 -0.224367 5 2.022633 6 -2.022633 7 -2.022633 8 -2.022633 9 -2.022633 10 2.471367
For example, when selecting a molecule with IDX=6, enter “6” in the [Set Base Molecule] field under the [Crystal Surface] tab.
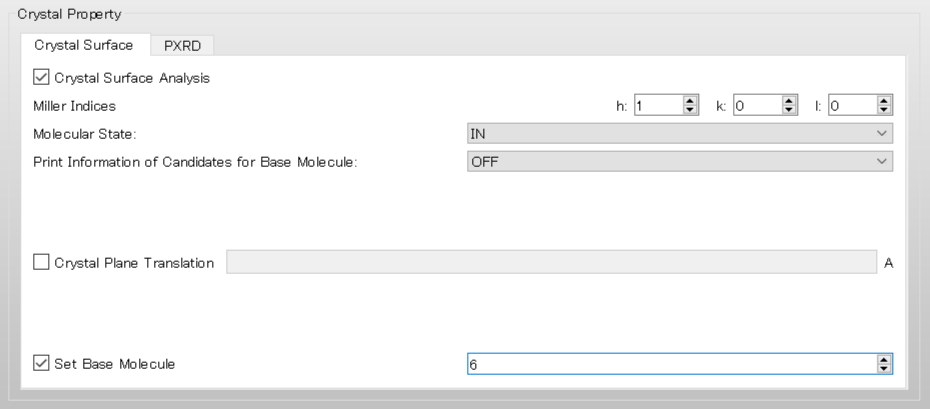
In the case of execution from the command line, add [CRYSTAL_PLANE_BASE=6] to the ini file and run the calculation.
tartronicacid.ini file
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(1,0,0) CRYSTAL_PLANE_BASE=6
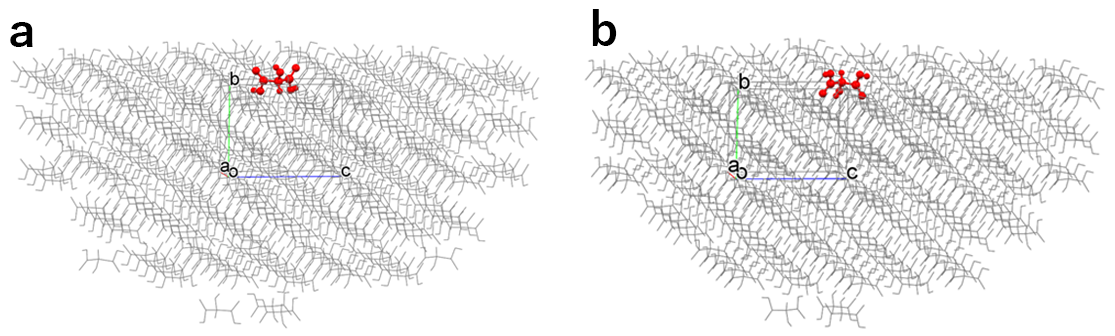
In the default setting, the hemispherical crystal model shown in the left figure is created, and the Emol is calculated. By setting “6” in [Set Base Molecule], the hemispherical crystal model shown in the right figure is created, and the Emol is calculated.
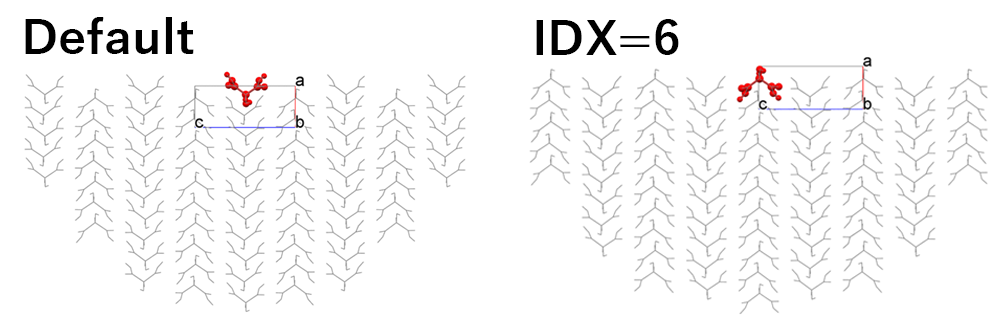
In the case of the left figure, the Emol is -10.73 kcal/mol.
In contrast, in the case of the right figure, the Emol is -34.15 kcal/mol.