Customize force field parameters
A molecular force field provides force field parameters necessary for evaluating various interactions. If the parameters required to calculate a target molecule are not available in the force field, the molecule cannot be calculated. This section explains how to add missing parameters and how to modify the parameters of force field in used. Calculations for the metal complex [Ti(H2O)6]3+ and acetic acid CH3COOH are shown below as example.
Metal complex [Ti(H2O)6]3+
Input structure and its data of [Ti(H2O)6]3+ are shown below. CONFLEX cannot perform any calculations on the structure due to the lack of force field parameters for Ti3+.
Input structure of [Ti(H2O)6]3+
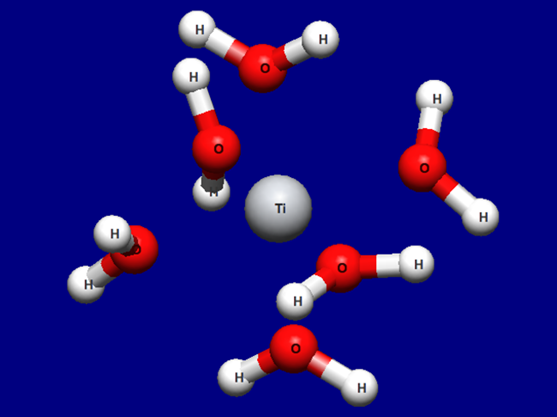
Structure data (Ti3+H2O6.mol)
Ti3+H2O6.mol
19 12 0 0 0 0 0 0 0 0 0 0
0.7679 -0.0417 0.0000 Ti 0 1 0 0 0 0 0 0 0 0 0 0
0.7679 1.9383 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.1801 2.4183 0.5879 H 0 0 0 0 0 0 0 0 0 0 0 0
2.7479 -0.0417 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
3.2279 -0.8728 -0.0216 H 0 0 0 0 0 0 0 0 0 0 0 0
0.7679 -0.0417 1.9800 O 0 0 0 0 0 0 0 0 0 0 0 0
0.1899 -0.6393 2.4600 H 0 0 0 0 0 0 0 0 0 0 0 0
0.7679 -2.0217 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.1945 -2.5017 -0.6020 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.2121 -0.0417 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.6921 0.0066 -0.8300 H 0 0 0 0 0 0 0 0 0 0 0 0
0.7679 -0.0417 -1.9800 O 0 0 0 0 0 0 0 0 0 0 0 0
1.3468 0.5551 -2.4600 H 0 0 0 0 0 0 0 0 0 0 0 0
0.1891 -0.6385 -2.4600 H 0 0 0 0 0 0 0 0 0 0 0 0
1.3558 2.4183 -0.5879 H 0 0 0 0 0 0 0 0 0 0 0 0
1.3414 -2.5017 0.6020 H 0 0 0 0 0 0 0 0 0 0 0 0
3.2279 0.7894 0.0216 H 0 0 0 0 0 0 0 0 0 0 0 0
1.3460 0.5558 2.4600 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.6921 -0.0901 0.8300 H 0 0 0 0 0 0 0 0 0 0 0 0
2 3 1 0 0 0 0
2 15 1 0 0 0 0
4 5 1 0 0 0 0
4 17 1 0 0 0 0
6 7 1 0 0 0 0
6 18 1 0 0 0 0
8 9 1 0 0 0 0
8 16 1 0 0 0 0
10 11 1 0 0 0 0
10 19 1 0 0 0 0
12 13 1 0 0 0 0
12 14 1 0 0 0 0
M END
[Execution from Interface]
Open the Ti3+H2O6.mol file using CONFLEX Interface.
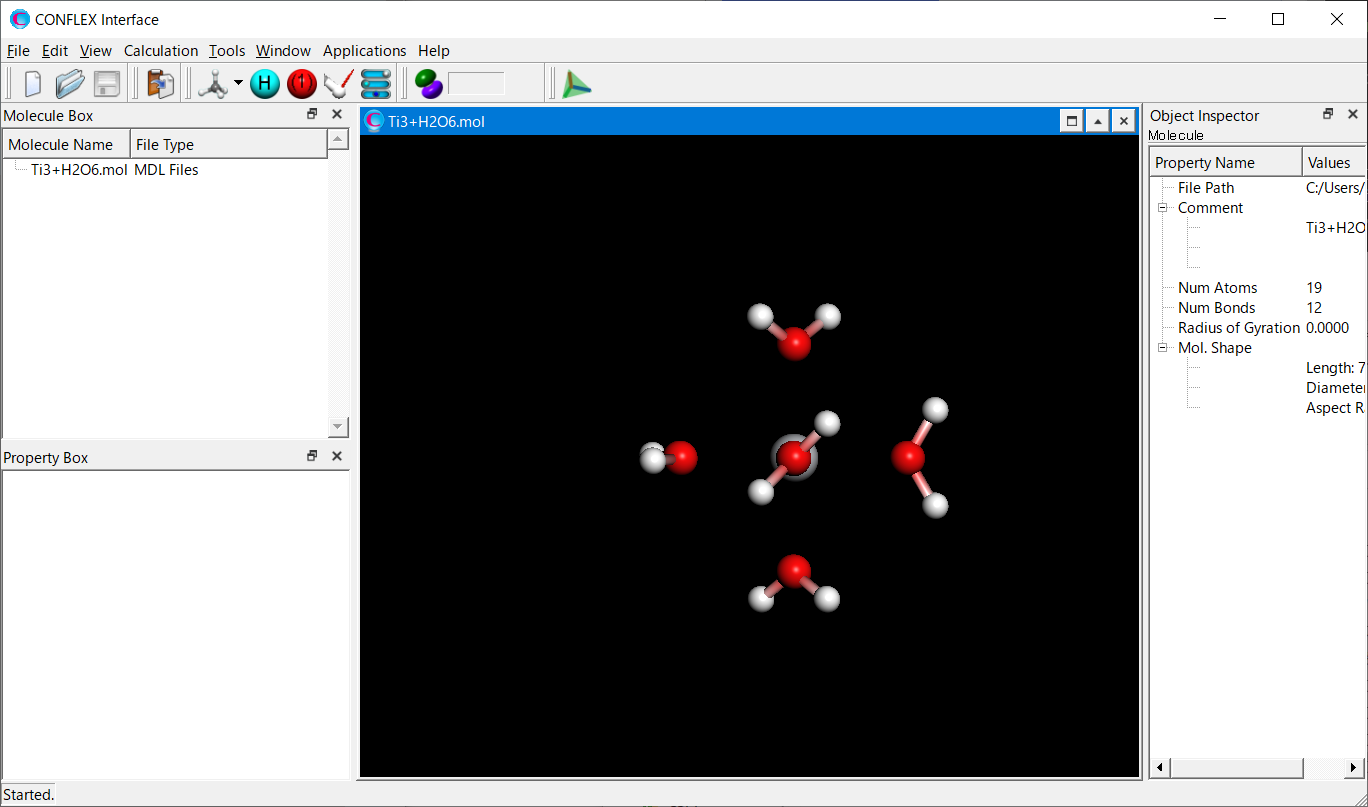
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. Next, click at the bottom right in the detailed settings dialog.
Add keywords of [SET_EXT_ATOM_TYPE=(1,900)], [VDWATOM=(900,-,0.45,6.0,4.0,1.4)], and [FRMCHG=(900,3,3.0)] to the dialog that appears.
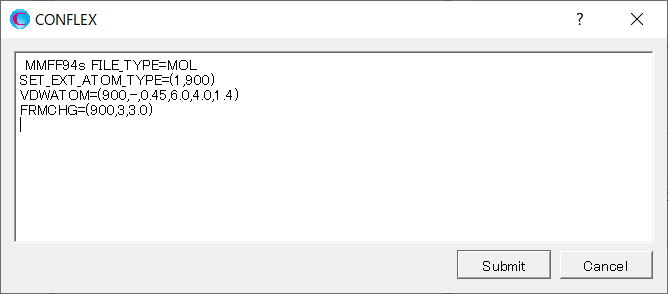
The [SET_EXT_ATOM_TYPE=(1,900)] sets the atom type of Ti3+ with serial number of 1 to 900.
The [VDWATOM=(900,-,0.45,6.0,4.0,1.4] sets the van der Waals interaction parameter for the atom with atom type 900.
The [FRMCHG=(900,3,3.0)] sets the formal charge of the atom with atom type 900 to +3 and its partial charge to +3.0, respectively.
After completing the calculation settings, click to start the calculation.
[Execution from command line]
The calculation settings are defined by specifying keywords in the Ti3+H2O6.ini file.
Ti3+H2O6.ini file
MMFF94S SET_EXT_ATOM_TYPE=(1,900) VDWATOM=(900,-,0.45,6.0,4.0,1.4) FRMCHG=(900,3,3.0)
The [SET_EXT_ATOM_TYPE=(1,900)] sets the atom type of Ti3+ with serial number of 1 to 900.
The [VDWATOM=(900,-,0.45,6.0,4.0,1.4] sets the van der Waals interaction parameter for the atom with atom type 900.
The [FRMCHG=(900,3,3.0)] sets the formal charge of the atom with atom type 900 to +3 and its partial charge to +3.0, respectively.
Store the Ti3+H2O6.mol and Ti3+H2O6.ini files in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Ti3+H2O6enter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].
Calculation results
The optimized structure is shown below. For how to visualize this, please refer to [Visualization of calculation results].
Optimized structure of [Ti(H2O)6]3+
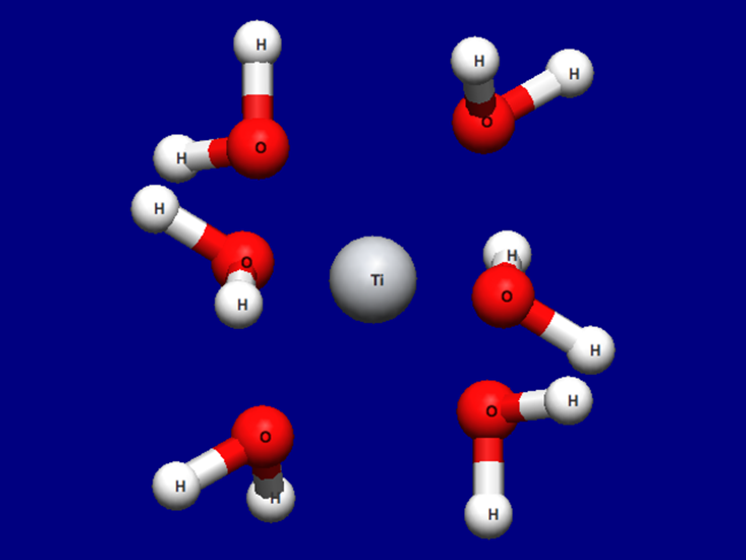
Structure data (Ti3+H2O6-F.mol)
Ti3+H2O6-F.mol
CONFLEX 14011413263D 1 1.00000 -477.60875 0
TH ,E = -477.609, G = 2.324E-07, M(0) MMFF94S(2010-12-04HG)
19 12 0 0 0 V2000
-0.0000 0.0000 0.0000 Ti 0 1 0 0 0
1.1603 0.8140 1.4174 O 0 0 0 0 0
1.9869 1.3938 1.4008 H 0 0 0 0 0
-1.1603 -0.8140 1.4174 O 0 0 0 0 0
-1.1501 -1.6932 1.9140 H 0 0 0 0 0
-1.1511 1.6410 0.0000 O 0 0 0 0 0
-1.9745 1.9212 -0.5131 H 0 0 0 0 0
-1.1603 -0.8140 -1.4174 O 0 0 0 0 0
-1.1468 -0.8045 -2.4271 H 0 0 0 0 0
1.1603 0.8140 -1.4174 O 0 0 0 0 0
1.9836 0.5051 -1.9140 H 0 0 0 0 0
1.1511 -1.6410 -0.0000 O 0 0 0 0 0
1.9745 -1.9212 0.5131 H 0 0 0 0 0
1.1344 -2.5106 -0.5131 H 0 0 0 0 0
1.1468 0.8045 2.4271 H 0 0 0 0 0
-1.9869 -1.3938 -1.4008 H 0 0 0 0 0
-1.9836 -0.5051 1.9140 H 0 0 0 0 0
-1.1344 2.5106 0.5131 H 0 0 0 0 0
1.1501 1.6932 -1.9140 H 0 0 0 0 0
2 3 1 0 0
2 15 1 0 0
4 5 1 0 0
4 17 1 0 0
6 7 1 0 0
6 18 1 0 0
8 9 1 0 0
8 16 1 0 0
10 11 1 0 0
10 19 1 0 0
12 13 1 0 0
12 14 1 0 0
M END
Acetic acid CH3COOH
Input structure and its data of CH3COOH are shown below.
Input structure of CH3COOH
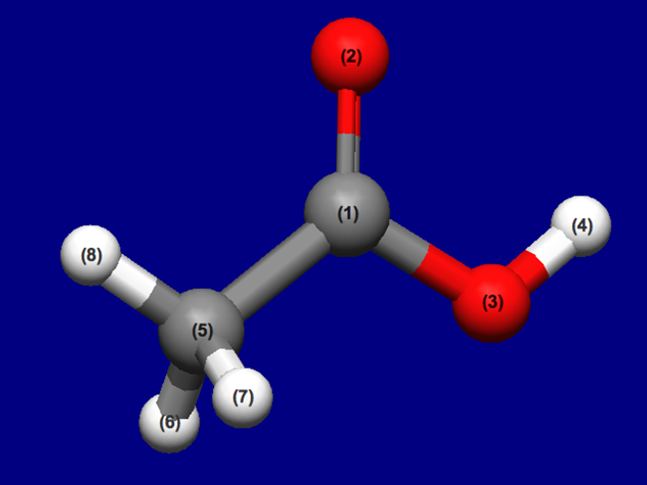
Structure data (CH3COOH.mol)
CH3COOH.mol
8 7 0 0 0 0 0 0 0 0 0 0
0.1268 0.0794 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.1528 0.7584 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.0615 0.7347 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.9219 1.6958 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.0496 -1.4504 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.5898 -1.7472 0.8747 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.5898 -1.7472 -0.8747 H 0 0 0 0 0 0 0 0 0 0 0 0
0.9117 -1.9203 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0 0 0 0
1 3 1 0 0 0 0
1 5 1 0 0 0 0
3 4 1 0 0 0 0
5 6 1 0 0 0 0
5 7 1 0 0 0 0
5 8 1 0 0 0 0
M END
The force field parameters for CH3COOH molecule are included in the force field provided by CONFLEX. However, you can modify these parameters using keywords.
[Execution from Interface]
Open the CH3COOH.mol file using CONFLEX Interface.
Select [CONFLEX] from the Calculation menu, and then click in the calculation setting dialog that appears. Next, click at the bottom right in the detailed settings dialog.
Add keywords for setting the force field parameters to the dialog that appears. About explanations of the keywords, please refer to the manual.
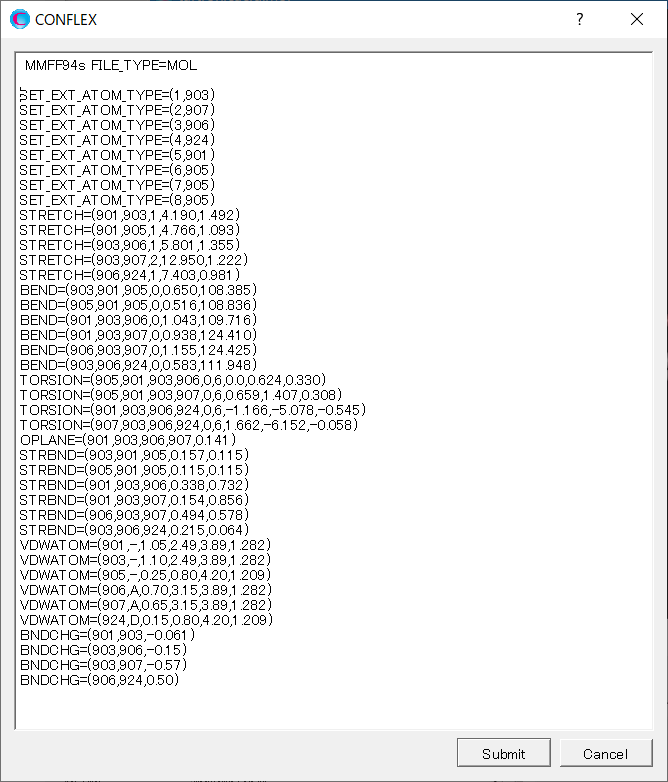
After completing the calculation settings, click to start the calculation.
[Execution from command line]
Add keywords for setting the force field parameters to the CH3COOH.ini file. About explanations of the keywords, please refer to the manual.
CH3COOH.ini file
MMFF94S SET_EXT_ATOM_TYPE=(1,903) SET_EXT_ATOM_TYPE=(2,907) SET_EXT_ATOM_TYPE=(3,906) SET_EXT_ATOM_TYPE=(4,924) SET_EXT_ATOM_TYPE=(5,901) SET_EXT_ATOM_TYPE=(6,905) SET_EXT_ATOM_TYPE=(7,905) SET_EXT_ATOM_TYPE=(8,905) STRETCH=(901,903,1,4.190,1.492) STRETCH=(901,905,1,4.766,1.093) STRETCH=(903,906,1,5.801,1.355) STRETCH=(903,907,2,12.950,1.222) STRETCH=(906,924,1,7.403,0.981) BEND=(903,901,905,0,0.650,108.385) BEND=(905,901,905,0,0.516,108.836) BEND=(901,903,906,0,1.043,109.716) BEND=(901,903,907,0,0.938,124.410) BEND=(906,903,907,0,1.155,124.425) BEND=(903,906,924,0,0.583,111.948) TORSION=(905,901,903,906,0,6,0.0,0.624,0.330) TORSION=(905,901,903,907,0,6,0.659,1.407,0.308) TORSION=(901,903,906,924,0,6,-1.166,-5.078,-0.545) TORSION=(907,903,906,924,0,6,1.662,-6.152,-0.058) OPLANE=(901,903,906,907,0.141) STRBND=(903,901,905,0.157,0.115) STRBND=(905,901,905,0.115,0.115) STRBND=(901,903,906,0.338,0.732) STRBND=(901,903,907,0.154,0.856) STRBND=(906,903,907,0.494,0.578) STRBND=(903,906,924,0.215,0.064) VDWATOM=(901,-,1.05,2.49,3.89,1.282) VDWATOM=(903,-,1.10,2.49,3.89,1.282) VDWATOM=(905,-,0.25,0.80,4.20,1.209) VDWATOM=(906,A,0.70,3.15,3.89,1.282) VDWATOM=(907,A,0.65,3.15,3.89,1.282) VDWATOM=(924,D,0.15,0.80,4.20,1.209) BNDCHG=(901,903,-0.061) BNDCHG=(903,906,-0.15) BNDCHG=(903,907,-0.57) BNDCHG=(906,924,0.50)
Store the CH3COOH.mol and CH3COOHl.ini files in a single folder, and execute the following command to start the calculation.
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par CH3COOHenter
The command above is for Windows OS. For other OS, please refer to [How to execute CONFLEX].