Conformation Search
Be sure to perform molecular geometry optimization before performing the conformation search.
In this section, we will use the file output from the molecular geometry optimization.
Copy the "C17H34-F.mol" file in the folder where you performed the structural optimization calculation in the previous step to a different folder.
First, let's assume that you have created a folder called "search" and copied the files into that folder.
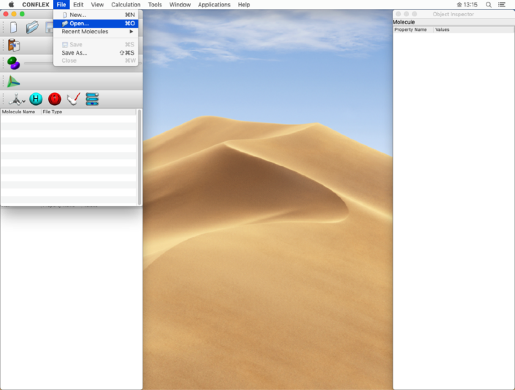
- From the "File" menu, click "Open" to open the "C17H34-F.mol" file in the "search" folder.
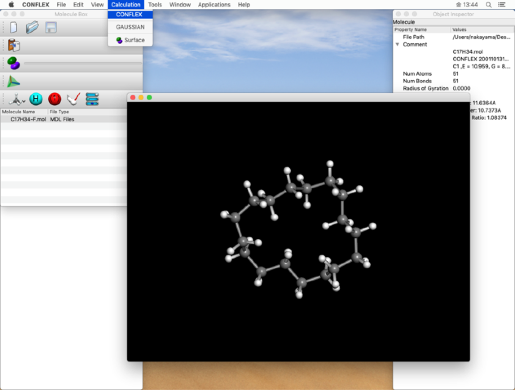
- Select "CONFLEX" from the "Calculation" menu to display the CONFLEX Settings dialog.
-
The calculation can be executed in the simple dialog, but you can also modify the settings in the detailed dialog and submit the job.
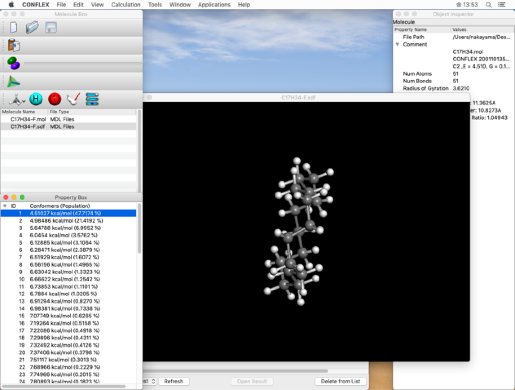
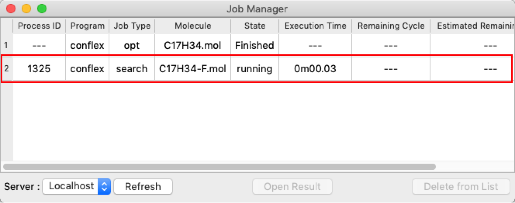
- When the job is executed, the "Job Manager" will be displayed. Make sure the "State" is "Finished" and double-click the row indicated by the red frame.
-
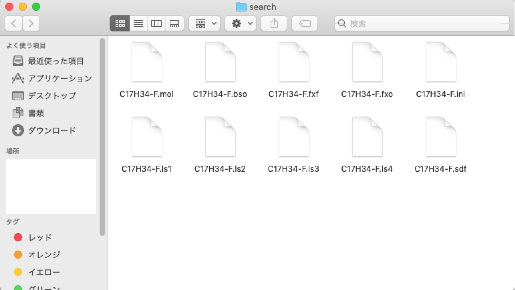
In the "search" folder, you will find "C17H34-F.mol" and nine other files:
- C17H34-F.bso
- C17H34-F.fxf
- C17H34-F.fxo
- C17H34-F.ini
- C17H34-F.ls1
- C17H34-F.ls2
- C17H34-F.ls3
- C17H34-F.ls4
- C17H34-F.sdf
For easy execution:
In the CONFLEX Settings dialog box, select "Conformation Search" from the "Calculation Type:" pull-down menu, and click "Submit" button.
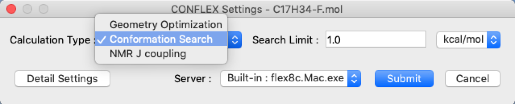
For Detail execution:
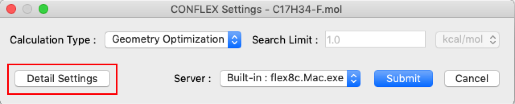
Click "Detail Settings" in the CONFLEX Settings dialog. The Detail Settings dialog box will appear.
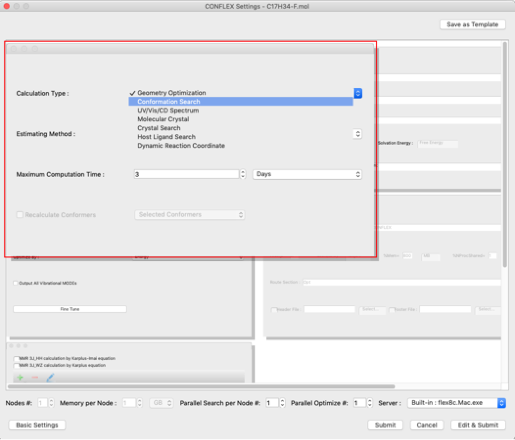
First, we will set the type of calculation. Select "Conformation Search" from the "Calculation Type:" pull-down menu in the "General Settings" dialog on the upper left.
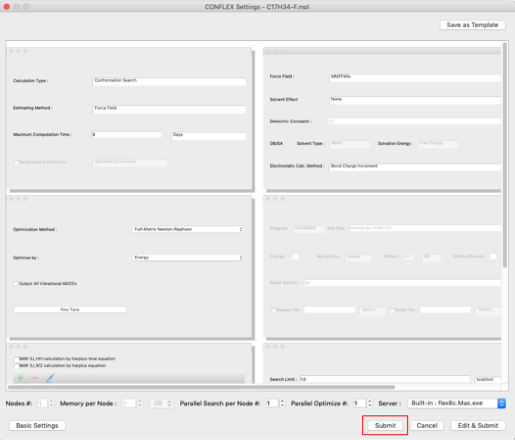
Click "Submit" to execute the conformation search job.
In the Detail settings dialog, you can also set the force field to be used, optimization method, search range, etc.
The molecule window, opened by double-clicking, displays the conformation isomer obtained by the search. Other conformational isomers are listed in the "Property Box" dialog. By clicking on an energy value, that conformational isomer will be displayed in the molecule window.