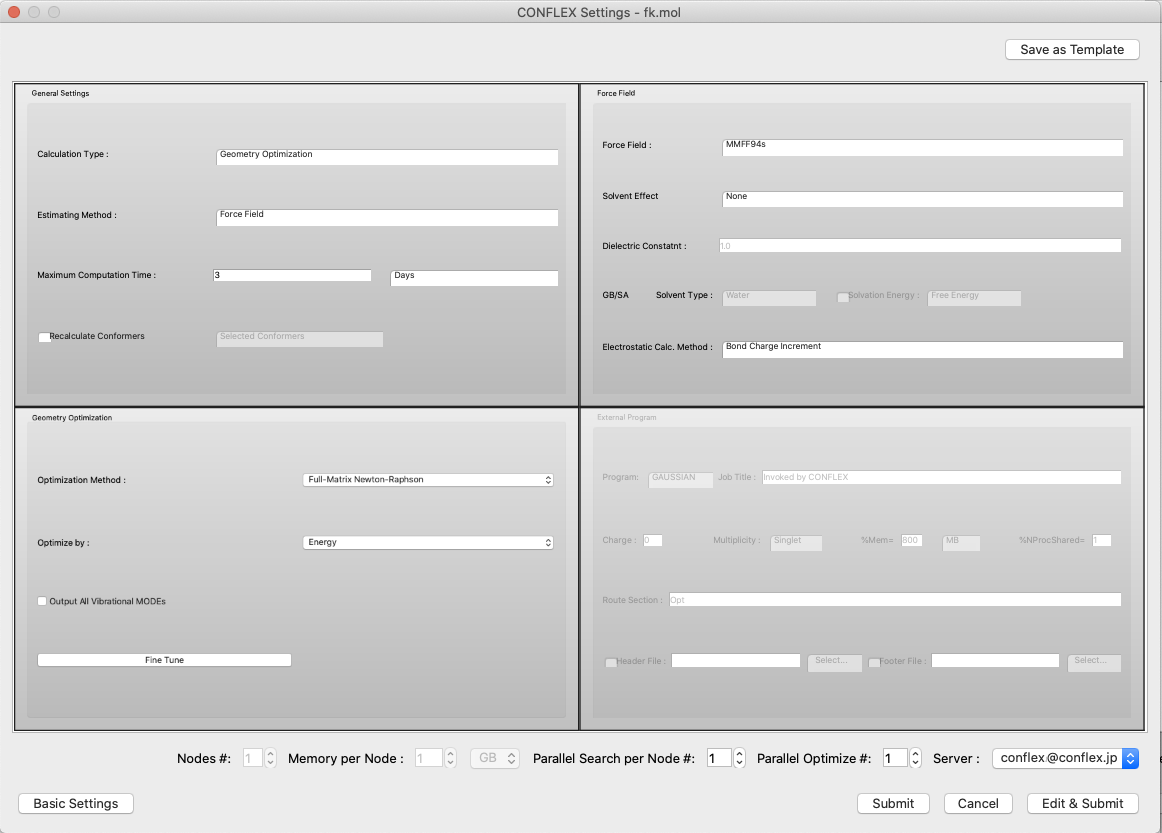
Detail Settings for CONFLEX Calculations
When the [Detail Settings] button in the initial dialog of the calculation settings is clicked, the detail setting dialog is displayed. Here, more advanced calculation settings can be configured. The dialog is an embedded form of each setting dialog, allowing you to see the entire dialog.
Click the "Basic Settings" button at the bottom left of the dialog to return to the original simple settings dialog.
- [Parallel Search #] and [Parallel Optimize #]
-
If you have the parallel version of CONFLEX, you can set a number greater than 1 in [Parallel Search #] and [Parallel Optimize #] to run parallel calculations.
The former will speed up the conformation search calculation, as each structure is optimized on a separate CPU during the conformation search.
The latter speeds up the optimization calculation of the structure by using multiple CPUs.
For normal molecules, it is more efficient to just set [Parallel Search #]. If the structure is large, such as a crystal, you can use both together to speed up the geometry optimization as well, which will speed up the whole process.
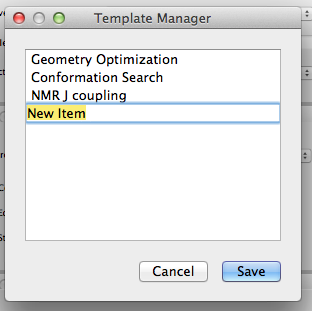
- Save as Template
-
Click the [Save as Template] button in the upper right corner of the dialog to save the current settings as a template. The following dialog will appear, and you can change “New Item” to any string you wish. These will be available for selection from the Simple Settings dialog.
You can also edit these settings by selecting “Scheme Editor” from the “Applications” menu and launching the editing program.
- Server:
- The location for carrying out the calculations is designated with [Server:].
- Submit
- When the [Submit] button is clicked, the job is submitted to the Job Manager.
- Edit & Submit
- When the [Edit & Submit] button is clicked, the text editor opens and the settings can be edited manually. This is commonly used to enter settings (keywords) not found in the dialog items list.
- In the main area of [CONFLEX Settings] dialog, the following sub-dialogs are embedded.
-
- General Settings
- Force Field
- Geometry Optimization
- Conformation Search
- Solvent Effect
- UV/Vis/CD Spectrum
- Crystal Optimization
- Crystal Search
- Host Ligand Search
- Dynamic Reaction Coordinate
- External Program
General Settings Dialog
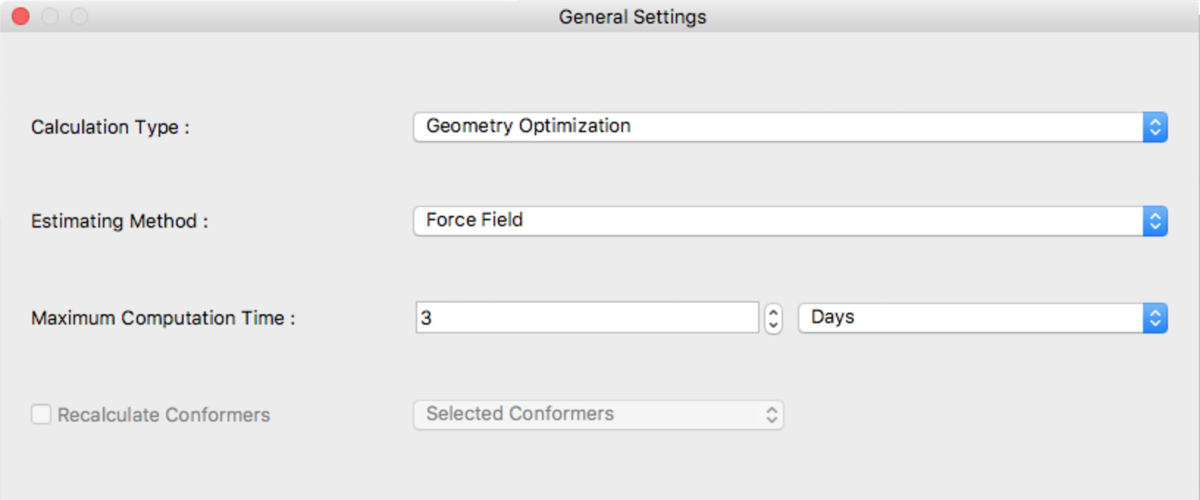
The type of calculation to be executed and time restrictions can be selected here.
Calculation Type:
The following are available for selection from Calculation Type:
- Geometry Optimization
- Structural optimization only.
- Conformation Search
- Conformational search + geometry optimization.
- NMR 3JHH - Coupling Constants
- Computation of proton 3JHH coupling constants.
- UV/Vis/CD Spectrum
- Simulates UV/Vis/CD Spectrum.
- Molecular Crystal
- Optimization of molecular structure, lattice constant, molecular orientation and translation position.
- Crystal Search
- Search and optimization of crystal structure.
- Host Ligand Search
- Host - Ligand Coordination search between more than one molecules.
- Dynamic Reaction Coordinate
- Simulates Dynamic Reaction Coordinate.
Estimating Method:
[Estimating Method:] specify the method used for geometry optimization. The following methods can be selected:
- Force FIeld
- Use force field
- External Program
- Invoke external program for geometry optimization (supports Gaussian program)
- Force FIeld and External Program
- Initially, optimize with force field, then optimize using external program (Gaussian)
Maximum Computation Time:
Maximum computation time is set here. The default unit of time is Days.
- Days
- Hours
- Minutes
Recalculate Conformers
Reoptimize conformers using CONFLEX on the calculation server. Mainly, used for UV/Vis/CD calculation. The range for reoptimizations are:
- Selected Coformers
- All Conformers
Force Field Dialog
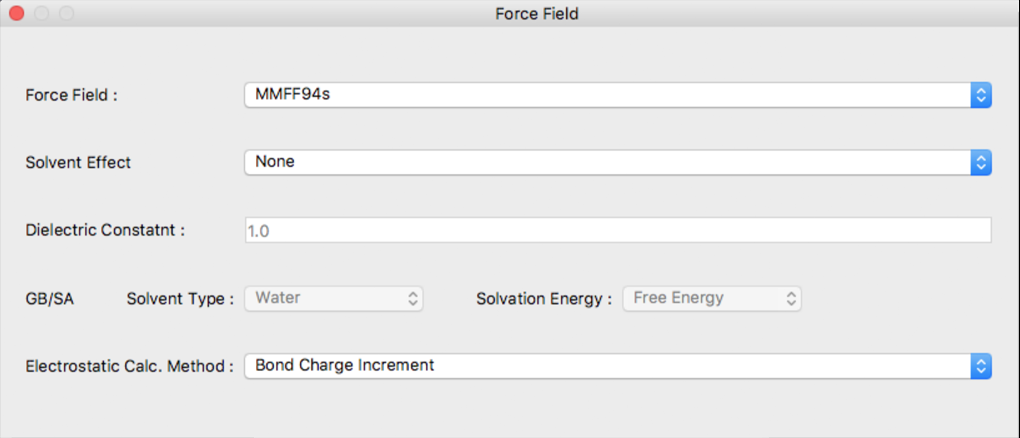
The setting for the force field used in structural optimization is configured in this dialog.
Force Field:
The force field used is designated using [Force Field:]. The choices are as follows:
- MMFF94s
- EMM2
Solvent Effect
Use of the solvent effect can be indicated in [Solvent Effect]. The choices are as follows:
- None
- solvent effect not used.
- Dielectric Constant
- specific dielectric constant is applied for electro- static interaction.
- GB/SA
- Generalized Born / Surface Area method is ap- plied; only MMFF94s
When [Dielectric Constant] is selected, a numerical value is set in the [Dielectric Constant:] box immediately below.
When [GB/SA] is selected, details can be set using the [Solvent Type:] and [Solvation Free Energy:] settings.
In [Solvent Type:], select the solvent type from the following:
- Water
- Octanol
In [Solvation Free Energy:], you can select the method of solvation energy analysis by GB/SA.
- Single Point
- Optimization
- Free Energy
For details, please see CONFLEX Keyword Options.
A method for calculating the electrostatic interaction can be selected using [Electrostatic Calc. Method:]
- Bond Charge Increment
- bond charge increment method
- NQEq
- new charge equilibrium method
- Bond Dipole
Geometry Optimization Dialog
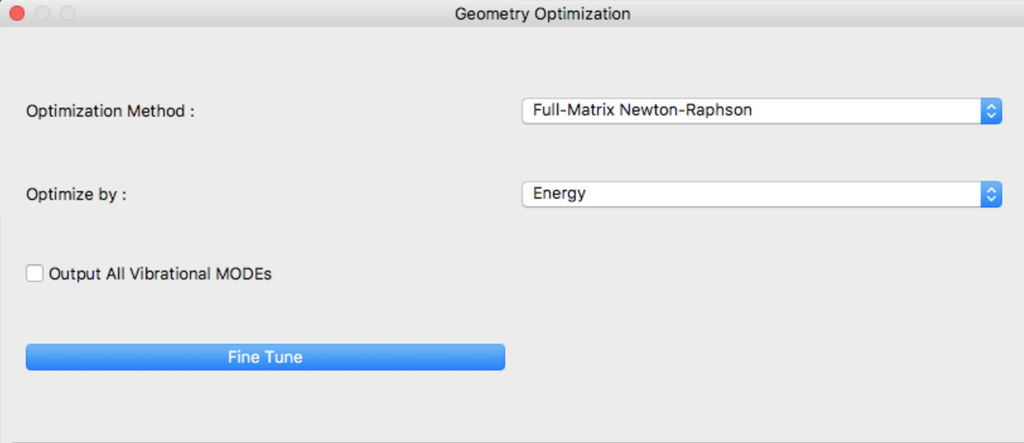
Settings for geometry optimization are made in this dialog.
Optimization Method:
An optimization algorithm is selected using [Optimization Method:] The [Full-Matrix Newton-Raphson] is the default and recommended method.
- Full-Matrix Newton-Raphson
- No Optimization
- Steepest Descent
- Conjugate Gradient
- Variable Metric
- FAST for large molecule
- PRECISE
- Frontier Mode Following
- Group Optimization
For details, please see the CONFLEX Manual
Optimize by:
Objective function to be minimized during geometry optimization is designated in [Optimize by:]
- Energy
- Gradient
Output All Vibrational MODEs
When [Output All Vibrational MODEs] is ticked, all of the vibrational moments are outputted.
Fine Tune
By clicking [Fine Tune] Button, dialog switch to Details Settings dialog.
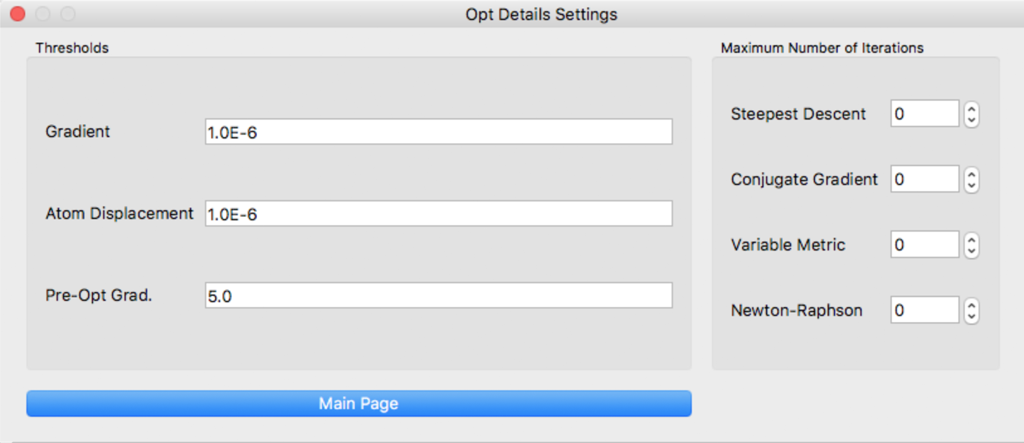
[Thresholds] defines several threshold values. [Maximum Number of Iterations] defines maximum number of iterations for each algorithm.
For details, please see the CONFLEX Manual
Conformation Search Dialog
The settings for conducting a conformational search are configured here.
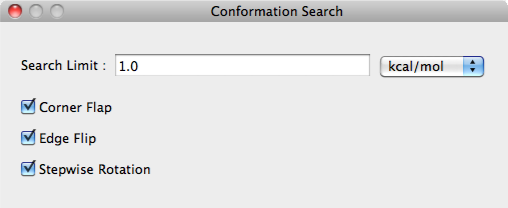
Search Limit:
Search parameters are designated in the [Search Limit:] box using the either kcal/mol or percentage (%).
Corner Flap, Edge Flip, Stepwise Rotation
The perturbation method for conformational search is selected in the [Corner Flap] [Edge Flip] [Stepwise Rota- tion] boxes.
Please see keyword manual for details.
UV/Vis/CD Spectrum Dialog
Specification of UV/Vis/CD Spectrum calculation is done by General Settings Dialog's [Calculation Type:]. In response to select [UV/Vis/CD Spectrum], the following dialog is displayed.
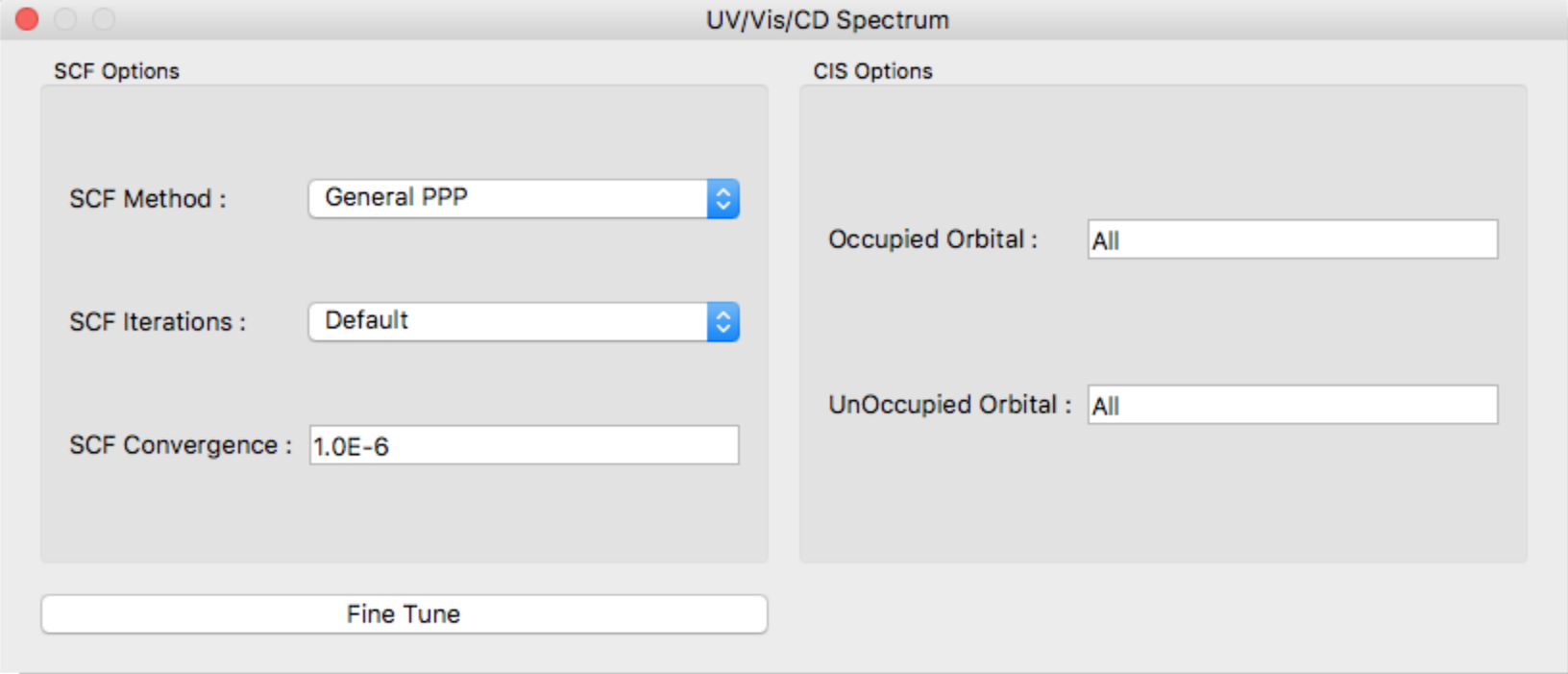
SCF Method:
In the [SCF Method:] field, specify the SCF calculation method. The options are as follows:
- GeneralPPP
- Perform general PPP/SCF-MO calculation.
- Variable Electronegativity
- Perform Allinger's Variable electronegativity method calculation.
SCF Iterations:
Specifies the number of maximum iterations (default: 50).
SCF Convergence:
Specifies the threshold values of SCF convergence.
Occupied Orbital, UnOccupied Orbital
Specifies number of occupied and unoccupied orbitals for CI single calculation. Each default values are “All” orbitals.
With a click of a [Fine Tune] button, dialog switches to details settings dialog.
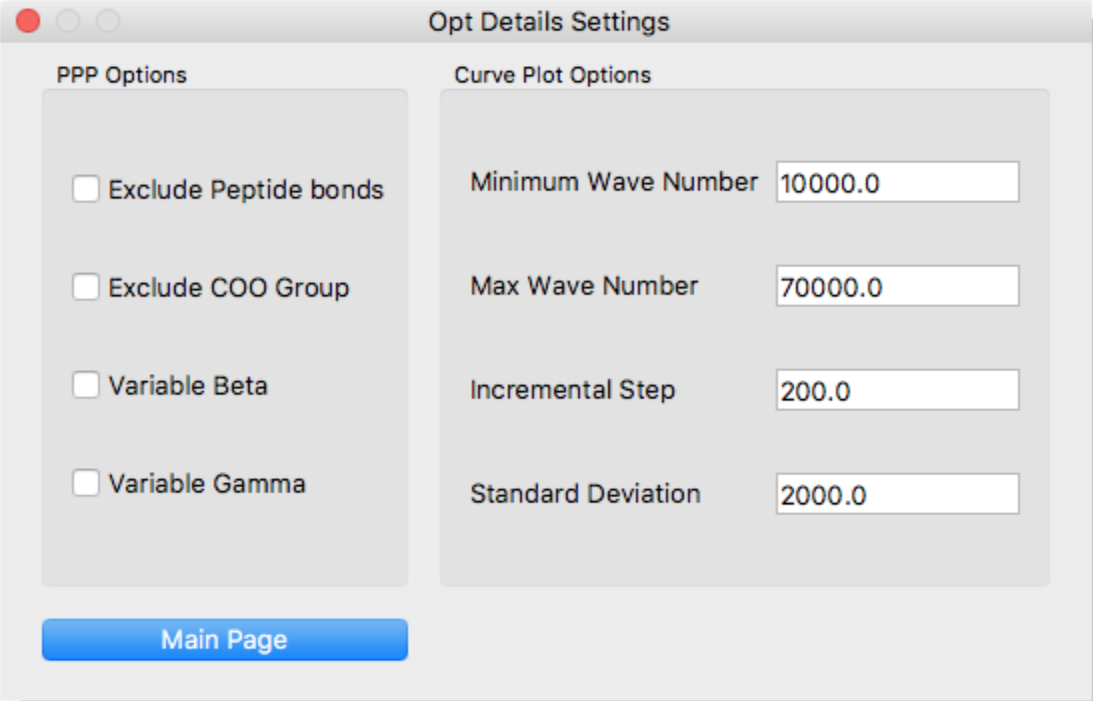
Configure the following here:
- Exclude Peptide bonds
- Exclude π-electrons of peptide bond.
- Exclude COO Group
- Exclude π-electrons of carboxyl group.
- Variable Beta
- Apply variable beta method.
- Variable Gamma
- Apply variable gamma method.
- Minimum Wave Number
- Starting point of spectrum based on the gauss approximation.
- Max Wave Number
- Terminal point of spectrum based on the gauss approximation.
- Incremental Step
- Incremental step width of spectrum.
- Standard Deviation
- Standard deviation of gauss distribution.
[Curve Plot Options]
Click the [Main Page] button at the bottom of the dialog to return to the original dialog.
NMR Dialog
Specify the 3J coupling constant calculation of NMR options.
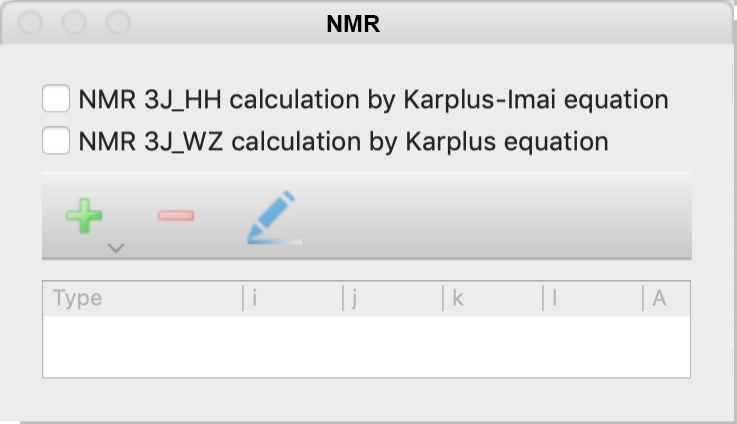
The options of the NMR calculation can be selected from the following.
- NMR 3J_HH calculation by Karplus-Imai equation
- 3JHH coupling constants around the Csp3-Csp3 bond are calculated using the Karplus-Imai formula.
- NMR 3J_WZ calculation by Karplus equation
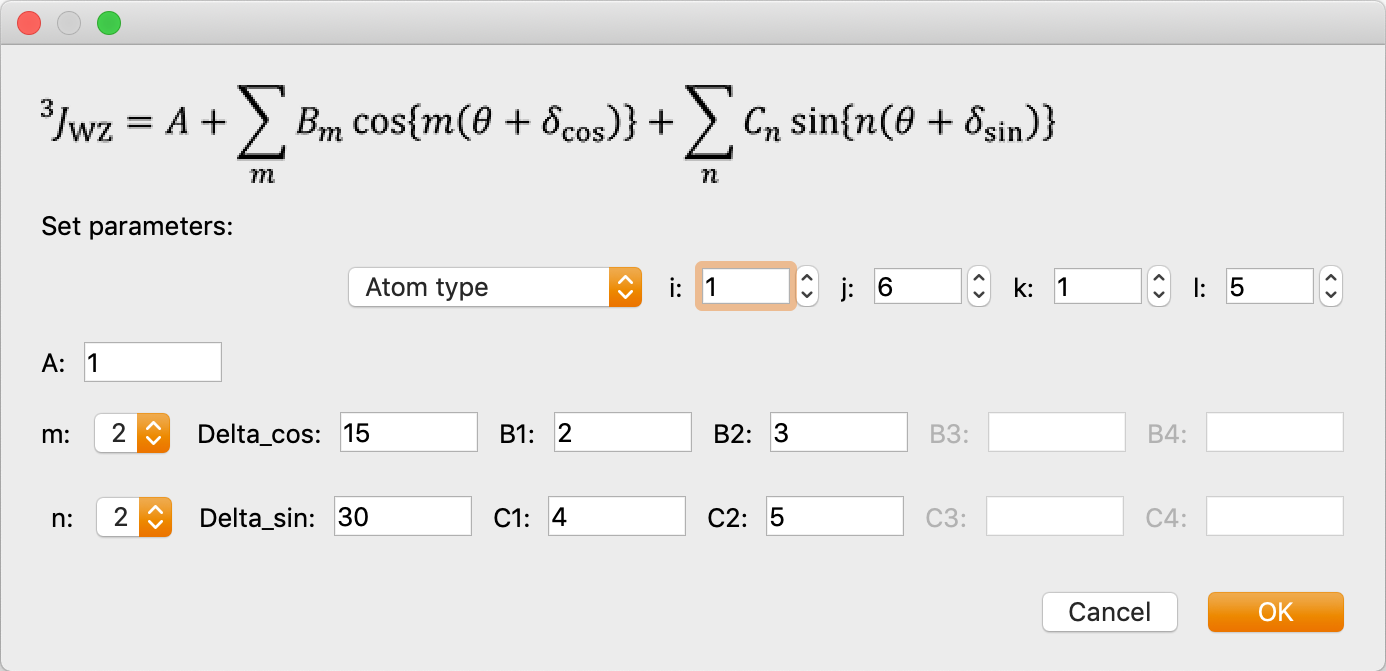
- Calculate the NMR 3Jcoupling constant around the atomic number I-J-K-L dihedral angle. When selected, a dialog box will appear as below, for entering parameters. Enter the desired parameters and click OK to display them in the table at the bottom of the original dialog. You can also click on the [+] or [-] icon in the dialog or the pencil icon to add, remove or modify previously entered parameters.
The formula is as follows:

Enter each parameter of this formula.
Procedure for Crystal Optimization
With the Molecular file for calculation already opened, perform the following steps:
- Select [Calculation]→[CONFLEX], and click the [Detail Settings] button.
- Select [Molecular Crystal] from the [Calculation Type] dropdown in the [General Settings] dialog box. [Crystal Optimization] and [Crystal Property] dialogs become visible.
-
Select the method of optimization from the Crystal Optimization dropdown in the Crystal Optimization dialog box.
Options for [Crystal Optimization:] are
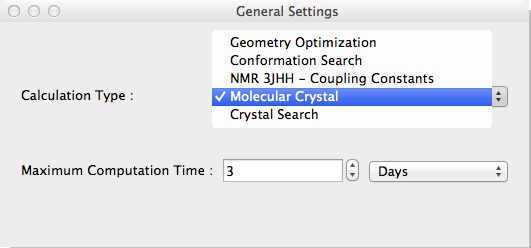
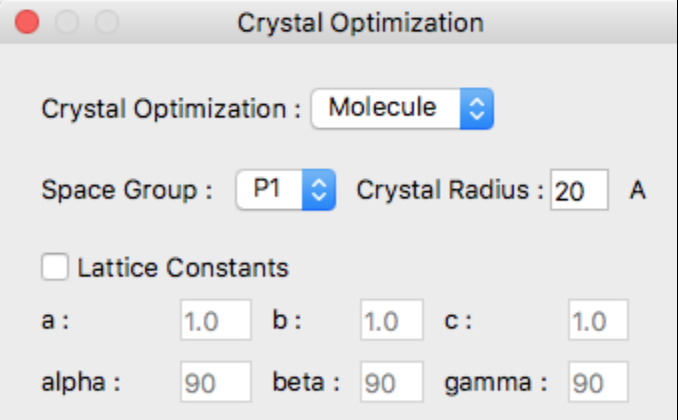
- Molecule
- The optimization method for calculating molecular geometry only.
- Rigid
- Lattice constants, molecular orientation and molecular translation will be optimized.
- All
- Referenced molecular structure, lattice constants, molecular orientation and molecular translation will be optimized.
- None
- Nothing optimized. Just output crystal properties.
If the input file is .cmf, or .cif and the information for space group and lattice constants is included, it will be displayed in the dialog box as well.
In this case, the [Lattice Constants] checkbox shown above will not be displayed.
If the input file does not contain crystal structure data, such as MDL-Mol format (.mol), the space group in the dialog will be displayed as [P1], and the lattice data a, b, and c will be displayed as 1.0, and alpha, beta, and gamma will be displayed as 90, respectively.
For the calculation, the initial values of the lattice constants will be set automatically, but if you want to specify them explicitly, check the [Lattice Constants] checkbox and enter the lattice constants.
Space Group:
The [Space Group] popup lists the typical space groups as follows:
- P1
- P-1
- P21/C
- P212121
- P21
- C2/C
- PBCA
- PNA21
- PNMA
- PBCN
If you want to specify other than these, select [Other...] item. You may enter the desired space group into the dialog box to open.
Crystal Radius:
You may specify the spherical crystal radius via the [Crysta Radius] text field.
If the calculation type is [Molecular Crystal], another dialog [Crystal Property] will be displayed.
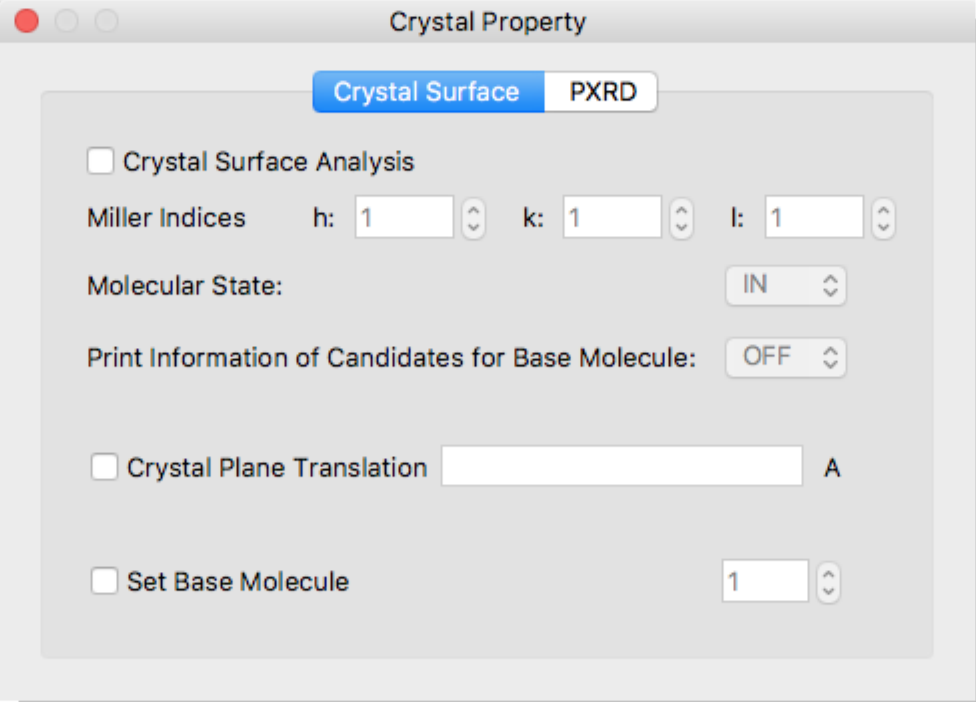
Using [Crystal Property] dialog box, you can specify details of the [Crystal Surface Analysis] or [Powder X-ray Diffraction] calculations.
Crystal Surface Tab
Crystal Surface Analysis
Check [Crystal Surface Analysis] to perform crystal surface analysis. The crystal surface is specified in the [h:], [k:], and [l:] boxes of [Miller Indices].
Molecular State:
Specifies a state of target molecule for energy calculation.
- IN
- The molecule “in” the crystal plane.
- ON
- The molecule “on” the crystal plane.
Print Information of Candidates for Base Molecule:
Information of candidates for a target molecule will be output to bso file.
Crystal Plane Translation
Specifies horizontal position of interface. If not given, this value is set automatically.
Set Base Molecule
Specifies a target molecule for energy calculation by molecule index. If not specified, the target molecule is set automatically.
PXRD
Click [PXRD] in the upper part of the [Crystal Property] dialog to switch to the setting window for powder X-ray diffraction pattern calculation.
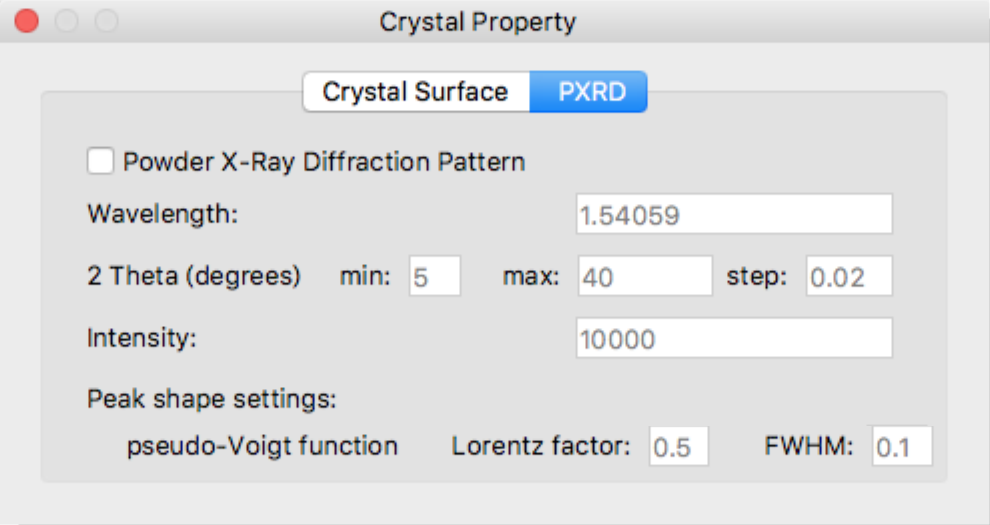
Powder X-Ray Diffraction Pattern
Define detail settings for Powder X-Ray Diffraction Pattern simulation.
Change the values of the various parameters as necessary.
- Wavelength
- Defines wavelength of radiation
- 2 Theta (degrees)
- Defines range of 2-theta and incremental step
- Intensity
- Defines highest intensity in the diffraction peaks
- pseudo-Voight function Lorentz factor
- Defines a value of Lorentz-polarization factor in the pseudo-voigt function
- pseudo-Voight function FWHM
- Defines a value of full width at half maximum in the pseudo-voigt function
[Peak shape settings:]
Crystal Search Calculation
To perform a crystal structure search, select [Crystal Search] from [Calculation Type:] in [General Settings].
If you want to change the optimization method of crystal structure, you can use [Crystal Optimization] in the [Crystal Optimization] dialog. If you want to change the method of crystal structure optimization, you can set it in [Crystal Optimization] in the [Crystal Optimization] dialog.
When the calculation settings are set to [Crystal Search], the [Fix Lattice Constants] checkbox appears in the [Crystal Optimization] dialog, allowing you to fix the lattice constants. When searching for a crystal structure, the initial value of the lattice constants is automatically set, but if you want to fix the lattice constants to a specific value, check the [Fix Lattice Constants] checkbox and specify the value of the lattice constants.
The following [Crystal Search] dialog will appear:
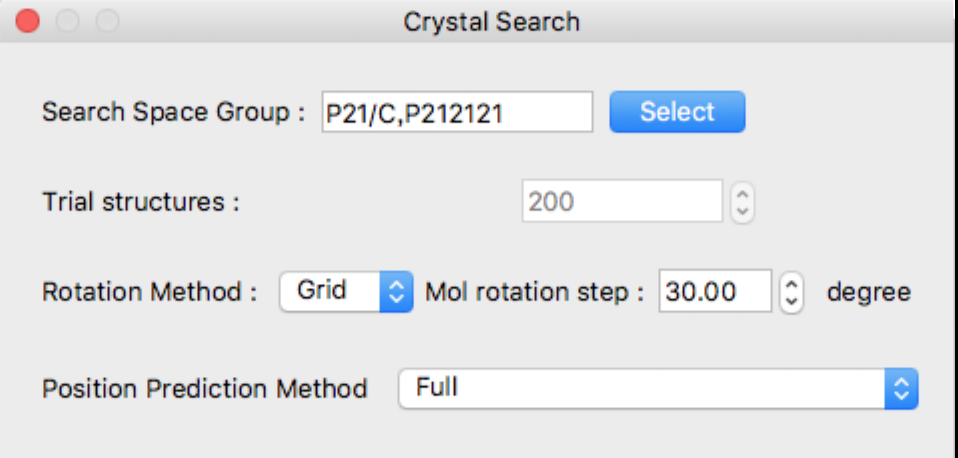 In the Crystal Search dialog, you can select the following:
In the Crystal Search dialog, you can select the following:
- Space group used for search
- Number of trial structures
- Method for rotating asymmetric units and step size
- How to find the translational position of an asymmetric unit
Space groups can also be selected by clicking the [Select] button to the right of the [Search Space Group] text box.
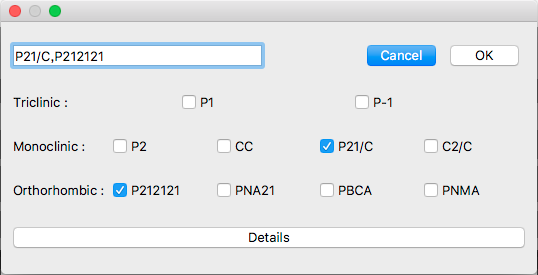
This dialog shows typical 10 space groups. If you want to select other space group(s), click [Details] button.
The detail dialog contains all space groups.
If you want to go back to original dialog box, click [Top 10] button.
Host-Ligand Coordination Search
Select [Host Ligand Search] from the [Calculation Type:] dropdown in the [General Settings] dialog box. [Host Ligand Search] dialog become visible.
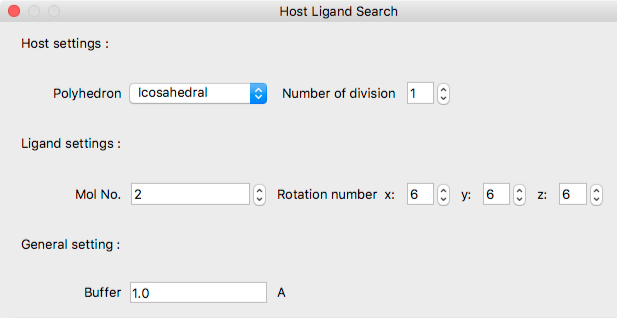
Host Settings:
Under Host settings:Polyhedron, select the polyhedron that defines the standard ligand molecular configuration around the host molecule. The choices are as follows:
- Tetrahedral
- Cube
- Octahedral
- Icosahedral
[Number of divisions] specifies the number of divisions for each face of the above polyhedron. Depending on the number specified here, the triangle will be divided at the midpoint of each edges, and the position of the ligand will be increased.
Ligand Settings:
- Mol No.
- Specifies ligand molecule number.
- Rotation Number
- Set the initial orientation of the ligand molecule rotating around x, y and z axes by 360/n degrees, respectively.
General settings:
- Buffer
- Distance (Å) between spheres surrounding both host and ligand molecules.
Dynamic Reaction Coordinate(DRC)
Select [Dynamic Reaction Coordinate] from the [Calculation Type:] dropdown in the [General Settings] dialog box. [Dynamic Reaction Coordinate] dialog become visible.
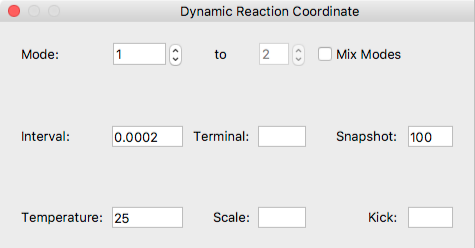
- Mode:
-
Specify one or multiple normal mode used for DRC calculation.
If you want to perform DRC calculation with more than one normal modes, check the [Mix Modes] box, then specify the range using [to] box. - Interval
- Specify the time step in ps.
- Terminal
- Speciify the termination time in ps.
- Snapshot
- Output the coordinate data every n steps.
- Temperature
- Set temperature in degrees centigrade.
- Scale
- Set scaling factor for amplitude of selected nomal mode(s).
- Kick
- Set value for scaling factor instead of sum of the frequencies in kcal/mol.
For details, see Tutorials.
Conformation Search Calculation invoking External Program (Gaussian 09 or 16)
Select the following options from [Estimating Method:] in the [General Settings] dialog box. In this case, external quantum chemistry program, Gaussian is called to optimize geometry instead of Molecular Force Field method.
- External Program
- Force Field and External Program
[External Program] dialog become visible.
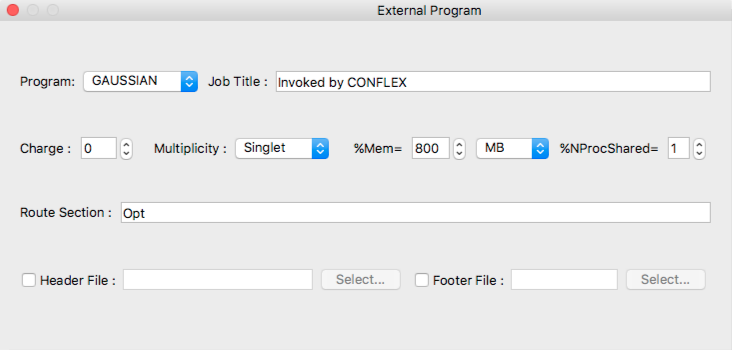
- Program:
- The current version of CONFLEX supports [GAUSSIAN] only as external program.
- Job Title
- Job title inserted to gaussian input file.
- Charge
- Specifies total charge of the whole molecule.
- Multiplicity
- Specifies spin multiplicity of the molecular system.
- %Mem=
- Specifies the amount of dynamic memory amount used by gaussian program. Unit is selected from popup list of:
- MB mega-bytes
- MW mega-worsd (N * 8 bytes)
- GB giga-bytes
- GW giga-words
- TB tera-bytes
- TW tera-words
- %NProcShared=
- Specifies the number of processors/cores on shared memory parallel execution on SMP multi-processors computer.
- Route Section
- Specifies the route section of gaussian job.
- Header File
- The gaussian settings can be specified using other file inside same folder of input file. After [Header File] box checked, you can specify [Header File] with [Select...] button.
- Footer File
- If gaussian input file needs footer settings, you should specify [Footer File]. After [Footer File] box checked, specify [Footer File] with [Select...] button.
For details, see Tutorials.