- Products
- CONFLEX Algorithm
CONFLEX explores the conformational space of flexible molecules and finds optimized structures of chemically important conformational isomers without exception.
Conventional structure optimization programs can only find local optimized structures that depend on the initial structure entered by the user. CONFLEX, on the other hand, prioritizes the creation of practically meaningful stable conformational isomers, thereby realizing efficient coordination space search.
Conformation Space Search
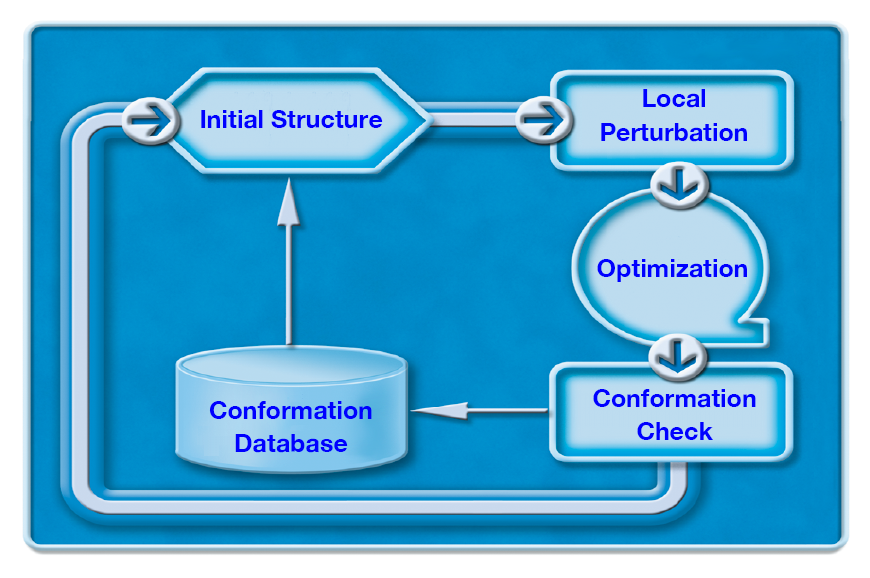
For convenience, the structures being processed are referred to differently depending on the particular stage of the search process:
- input structure is the input at the beginning of a search
- initial structure is called from the conformer storage database at the beginning of each perturbation cycle
- starting (trial) structure is the perturbed structure before geometry-optimization
- optimized structure is the structure after geometry-optimization
- stored structure is the optimized structure that survived the redundancy test, and it is saved in the conformer storage.
The algorithms for searching conformational space or a torsional hypersurface involve repeated sampling from the vast conformational space. The process, which is illustrated below (a curved arrow indicates a looping subprocess), has the following steps:
- Generation of an appropriate starting structure
- Geometry-optimization of the starting structure
- Comparison of the optimized structure with the stored conformers
Finally, a structure passing all comparisons is added to the list of stored conformers.
In the advanced conformational space search algorithms, the first step consists of two subprocesses:
- Selection of an initial structure from among the stored conformers
- Assignment of appropriate structural perturbation to the initial structure to produce the coordinates of a starting structure.
Whereas the overall scheme is similar to most of the known conformation-generators, new strategies have been implemented for each step. In the following sections, these strategies are described.
Reservoir Algorithm
The lowest-energy structure of the unused, stored conformers is chosen as the initial structure (a structure is considered to be unused if it has never been perturbed to generate a starting structure). This strategy is key to searching through the region in the conformational space, in the most efficient way towards a lower energy area. This is similar to a stream filling an empty reservoir by finding the the lowest point.
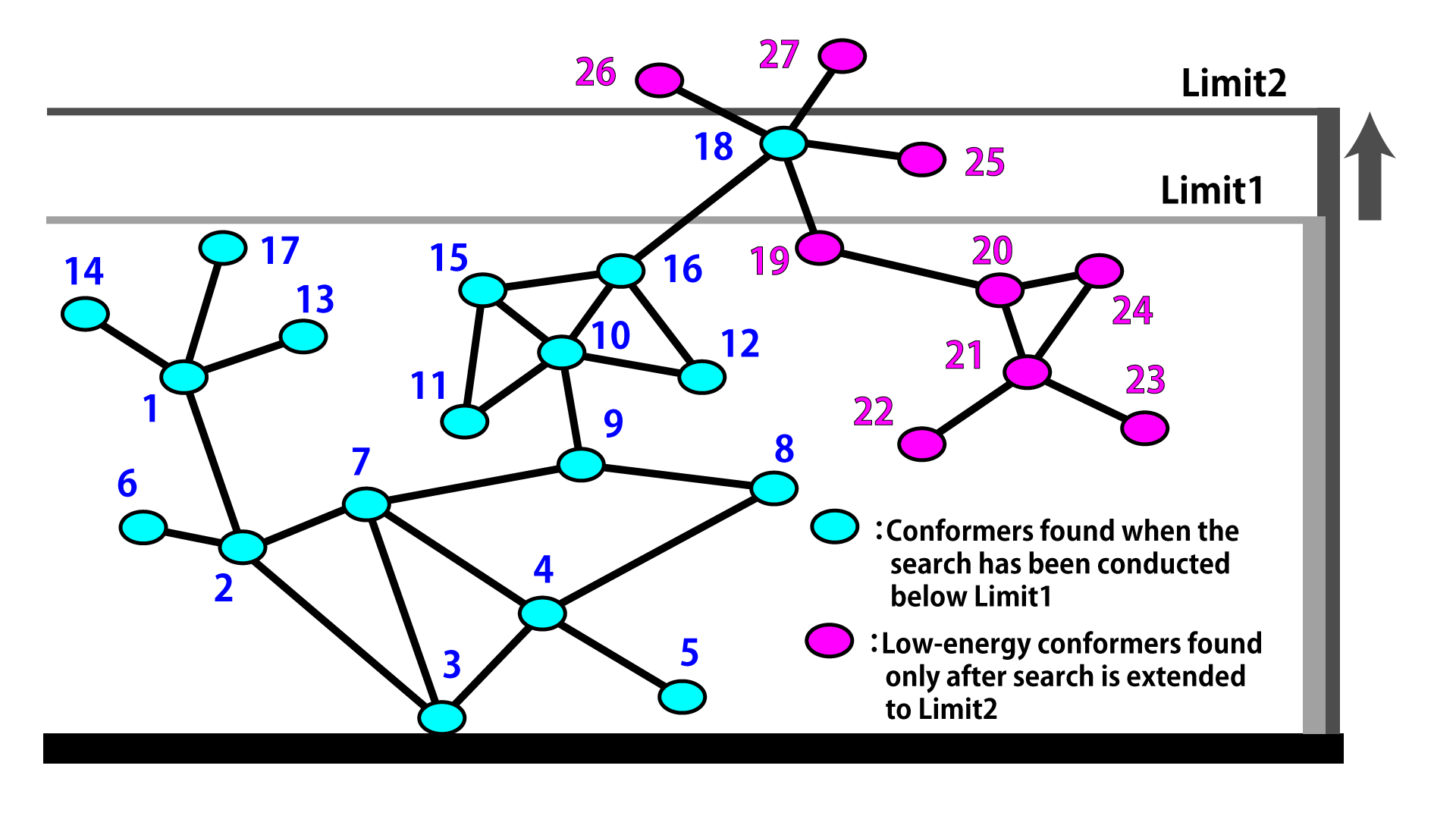
The strategy is illustrated schematically here for a conformational space search by the reservoir-filling algorithm.
- The initial structures are sampled in the order indicated by the number appearing next to the conformers (ellipses). A pair of conformers connected by a line comprise an initial structure and a conformer generated from that initial structure through perturbation and geometry-optimization.
- When the search limit is set to the level of Limit 1, the search is complete after the initial structures 1 through 17 have been processed.
- Then the search limit is increased to Limit 2, and conformers 18 through 25 are subjected to initial structures.
Variable Search Limit
When the global minimum or the instant global minimum in a local area is reached, the search moves towards a higher-energy area, and sometime local dips are overlooked in the process. The gradual expansion can be utilized to guarantee the thoroughness of the low-energy search.
The search limit is first decided - for example 5 kcal/mol from the global energy minimum. All the conformers within this first window (Limit 1) are used as the initial structures. During this search, all the conformers are saved, even those with energies higher than Limit 1.
The limit is increased to Limit 2 and all the conformers in the new window (between Limit 1 and 2) are subjected to initial structures in order of increasing energy. If local networks of conformers (LAN-a and LAN-b) are not connected within the first window, LAN-b is searched after LAN-c in the second window. A window width of 7 to 10 kcal/mol from the global minimum is generally sufficient to achieve an exhaustive search of the first, chemically meaningful window. Therefore, the variable limitation technique is a useful strategy for keeping the search region under control during the universal search.
Local Perturbation
If the perturbation cannot move the starting structure from the territory to which it initially belonged, subsequent geometry-optimization will return it to the same structure. The territories of similar conformers may be located in close vicinity in the conformational space, constituting a local network of local territories, and less similar conformers may be considered to belong to different localities. Therefore, the method of perturbing an initial structure to produce a new candidate conformer is also responsible for the efficiency of search.
To ensure exhaustive generation of all possible starting structures, local perturbation is applied to every flexible part in the initial structure. The following three modes of perturbation are designed to mimic the elementary process in the thermal movements of a molecule undergoing conformational change: corner flap and edge flip for endocyclic parts, and stepwise rotation for acyclic parts.
Corner Flap
Corner flap exploits the puckered feature of a ring structure, and involves the movement of a corner ring-atom to the other side of the local average plane. Two to four contiguous dihedral angles are simultaneously changed along the ring. The advantages of corner flap are:
- it is highly efficient in producing a new energy-minimum
- it can be considered to mimic a barrier-crossing step in the elementary process of thermal conformational interconversion
- it generally does not propagate itself, and can therefore be applied to every ring-atom. In many cases, as many starting structures as the number of ring-atoms can be obtained.
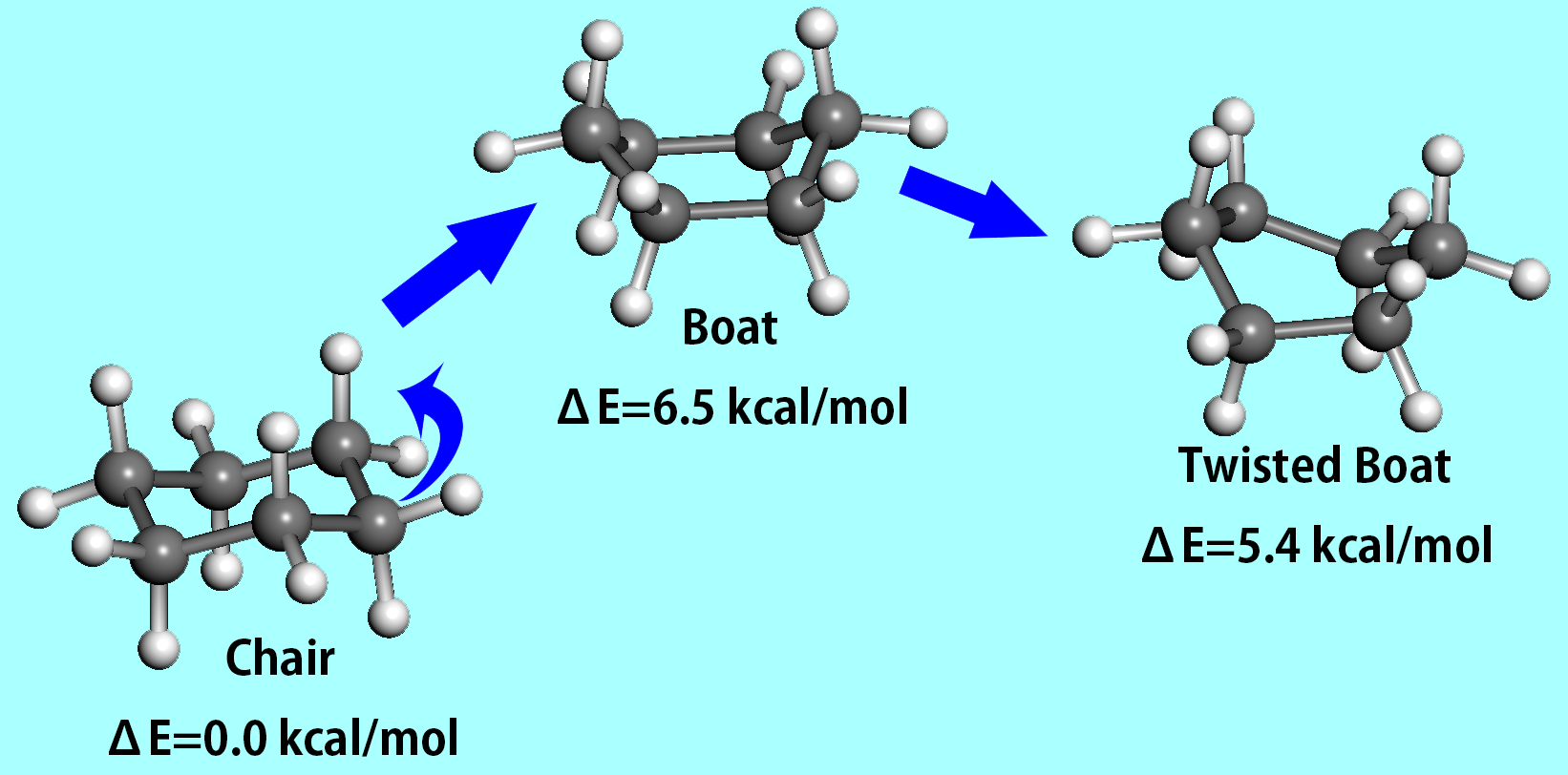
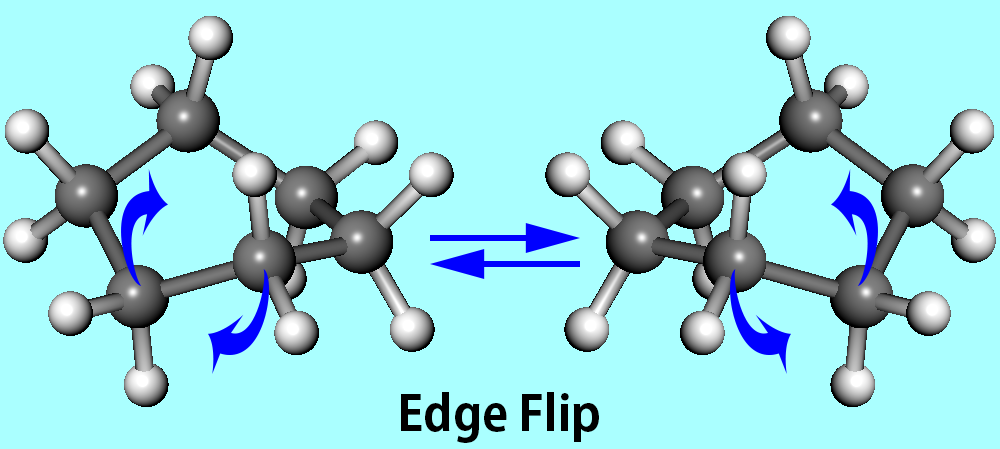
Edge Flip
Whilst corner flap is efficient for small to medium ring structures, it is sometimes an unsuccessful technique for larger rings. For example, a ring-atom that is lying on the average local ring plane and flanked by a pair of gauche bonds of the same sign cannot be flapped. Corner flap alone cannot produce all the nearby energy minima for larger rings where mechanisms of conformational interconversion involving more than four contiguous bonds exist.
Edge flip is where two adjacent ring-atoms are simultaneously given the corner flap in opposite directions, where a ring bond is flipped. Edge flip is best illustrated by the chair-to-twist boat conversion of cyclohexane. An edge flip perturbation mode, which proved to be effective for large rings, consists of simultaneous small flapping of two adjacent ring-atoms towards the inside of the ring. In contrast to the other two perturbation methods, both directed towards the outside of the ring, this mode is a local inflection occurring for ring structures with a large cavity space. The edge flip involves rotation of two to five contiguous ring bonds, and are called when certain combinations of three contiguous dihedral angles patterns (A, G, G') appear along the ring. Careful structural adjustments are given in order to ensure smooth transformation of local structure by edge flip, which is actually a much larger perturbation.
Stepwise Rotation
The acyclic part of a molecule is perturbed by stepwise rotation. Typically, a Csp3-Csp3 bond is given 120 and -120 rotations to produce a pair of new rotamers. This method, combined with the reservoir-filling strategy for guiding the search direction, searches low-energy conformers for short side chains and linear molecules with up to six to ten rotatable acyclic bonds. However, it is not as effective for large molecules with multiple branching, where unexpectedly high-energy starting structures are produced.
CONFLEX's MMFF
In the force fields included in CONFLEX, MMFF94 and MMFF94s use the “bond charge increment method” to place charges on most of the atoms in the molecule. These charges are parameterized to reproduce the charge distributions obtained from high-precision ab initio molecular orbital calculations on small molecules, but after the charge distributions are determined at the initial stage of the calculation, the charge values are kept fixed during optimization.
MMFF/NQEq, on the other hand, uses the “charge equilibrium method (QEq method)” instead of the above method to calculate the charge and electrostatic interaction energy.
In the QEq method, the charge distribution depends on the structure, so we have to recalculate the charge everytime the structure changes. However, the calculation itself is simple, so the increase in calculation time is very small, and the parameters are optimized by us, so we can calculate the electrostatic interaction with high accuracy in the same calculation time as other force fields.
Parallel CONFLEX
In CONFLEX, the initial structure is deformed by the rotation of the bonds in the chain and the flap/flip of the atoms that make up the ring. In Parallel CONFLEX, the structure optimization is performed on multiple CPUs/core. Then, the optimized structures obtained from each calculation are aggregated and checked for conformation.
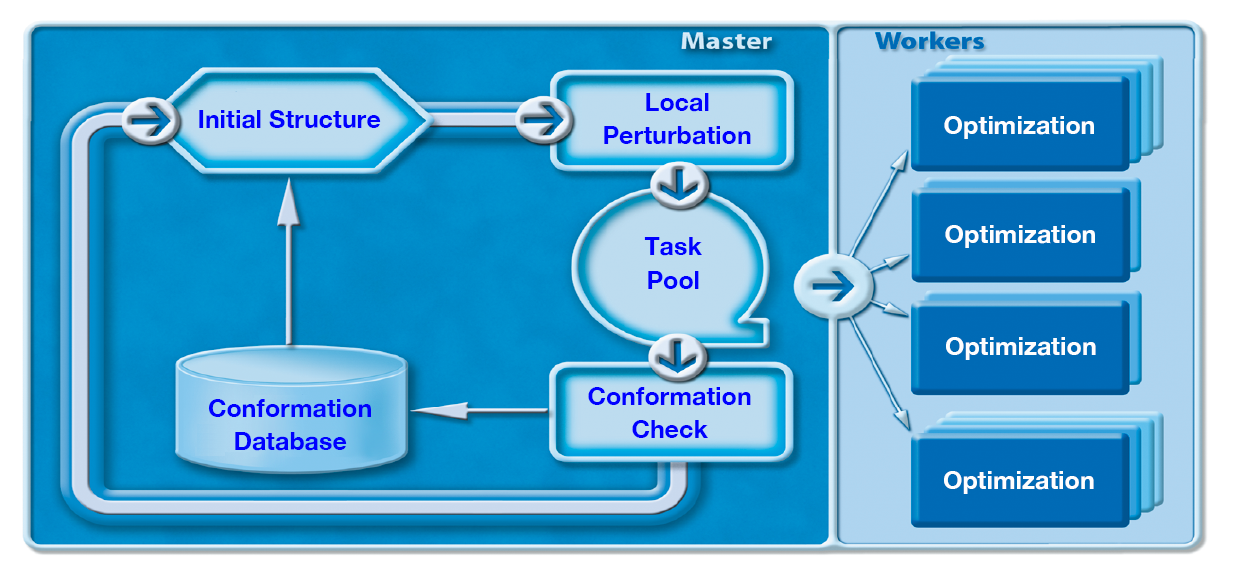
Significance of Parallelization
In particular, in the case of molecular crystal structure search, it is necessary to perform structural optimization of crystal polymorphs of various crystal structures. For those of you who analyze these structures by molecular calculations, Parallel CONFLEX will be effective in shortening your research time.
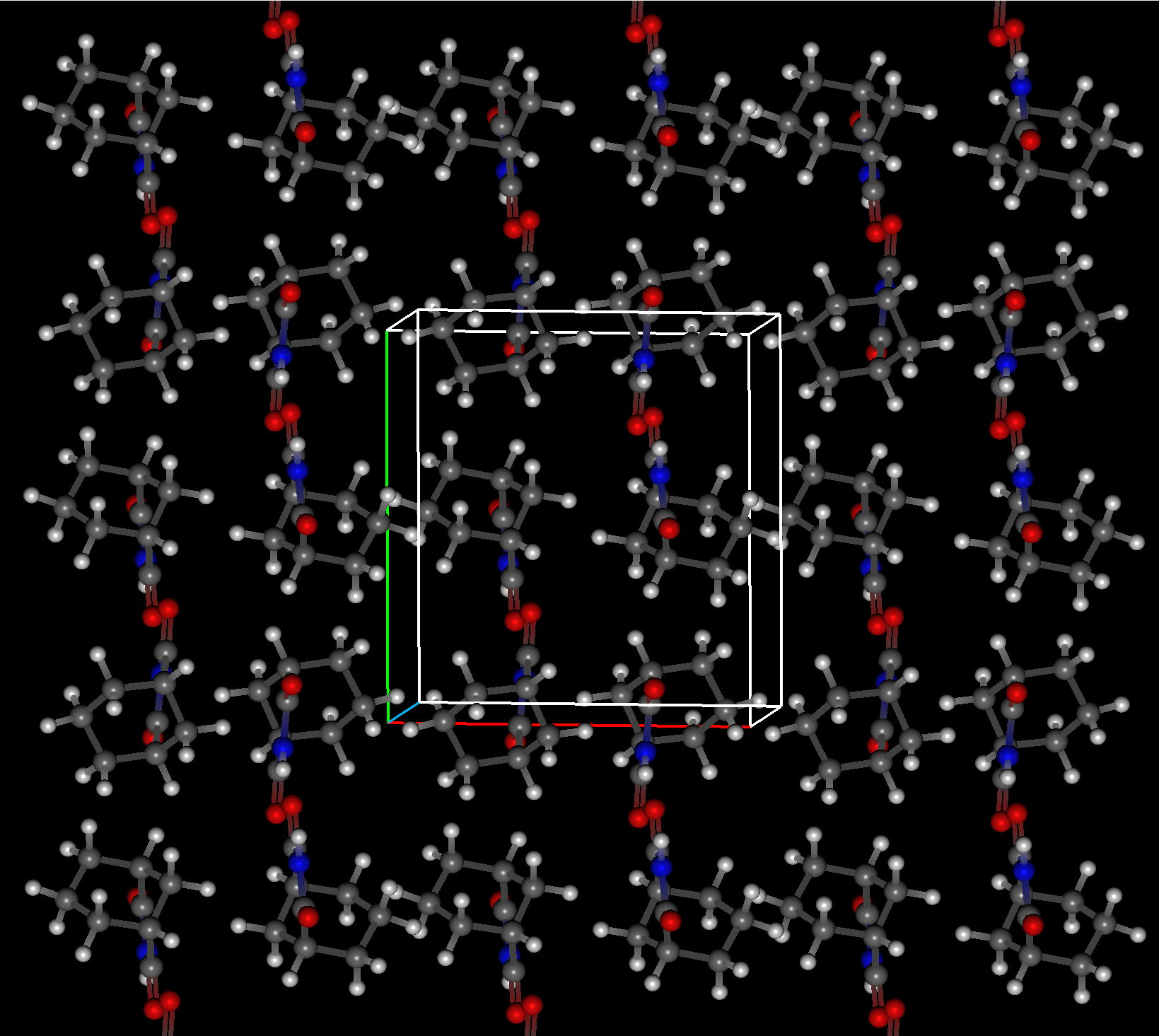
Crystal Structure Prediction of 3-aza-bicyclo(3.3.1)nonane-2,4-dione
For this molecule, CONFLEX generated 11,664 candidate crystal polymorph structures and optimized each of them to find 512 unique crystal polymorphs. The execution time is shown in the graph, and increasing the number of Workers efficiently reduces the execution time.