- Products
- What's NEW
New Features in CONFLEX 10

- Added "In-Situ Orientation Search" Feature (Basic, Pro) This feature allows searching for stable orientations of a specified molecule in systems containing multiple molecules, based on the arrangement in the input structure.
- Improved and Extended Output Files When Using PDB Files (Basic, Pro) When performing structure optimization or conformational search using a PDB file as input, the output PDB file now includes energy values. Additionally, mol and sdf files (for fewer than 1000 atoms) or mol2 files (for 1000 atoms or more) are now also generated.
- Additional Recognizable Amino Acid Residue Names (Basic, Pro) Added support for recognizing additional amino acid residue names when reading PDB files: HIE, HID, HIP, ASH, GLH, LYN.
- Enhanced Crystal Calculation Features The crystal calculation methods have been updated with new features such as molecular orientation control using quaternions and lattice deformation using the strain tensor.
- System Requirements
Windows (64bit): 10, 11
macOS: 13, 14, 15
Linux CONFLEX: RedHat EL 8.6, 8.8, 8.10, 9.2, 9.4, Ubuntu 22.04
(Please contact us for support on other Linux distributions)
As a result, stress tensor calculations are now supported, and crystal structure optimization calculations have been accelerated.
New Features in CONFLEX Interface 10

- Added Interface for CONFLEX DOCK Added functionality to submit and manage jobs for the new product "CONFLEX DOCK".
- Support for Loading Output Structures from CONFLEX DOCK Added support for reading PDB and Mol2 files output by CONFLEX DOCK.
- Expanded Multithreading for File Loading Enabled multithreaded loading of large files such as PDB files, allowing already loaded structures to be displayed while loading continues.
- Interface for Orientation Search Feature Implemented an interface for the new orientation search feature, allowing necessary settings for calculations to be configured.
-
Bug Fixes
- Illegal Character Check Implemented error display for illegal characters in filenames and paths. This helps avoid issues during job submission or monitoring caused by such characters.
- Revised Icons Updated program icons to improve visibility, including in dark mode.
- Amber File Bug Fix Fixed an issue where filenames were not displayed in the Job Manager when opening Amber files.
- Mol2 File Bug Fix Fixed a display issue for certain elements in Mol2 files.
- Revised Communication Protocol Revised the communication protocol to address potential delays when accessing external Linux servers for job-related operations.
- System Requirements
Windows (64bit): 10, 11
macOS: 14, 15
Linux CONFLEX: RedHat EL 8.6, 8.8, 8.10, 9.2, 9.4, Ubuntu 22.04
New Features in CONFLEX 9
- Addition of feature for crystal calculation (Pro)
- Pseudo forces generated by harmonic potentials can now be applied between molecules in an asymmetric unit.
The molecular arrangement subjected to the pseudo forces is constrained during a structure optimization.
- Pseudo forces generated by harmonic potentials can now be applied between molecules in an asymmetric unit.
- Correcting bond order (Basic, Pro)
- The keyword for correcting bond order between atoms that the valence is not fulfilled is added (except .pdb).
- Extension of Invoking Gaussian (Pro)
- The fixed atoms during optimization are reflected in Gaussian input file with setting CONFLEX keywords for partial geometry optimization.
- The connectivity of atoms can be added to Gaussian input file.
- Hardware Requirements: Newly supported platform from CONFLEX 9 Rev.C are highlighted in red.
- Windows (64bit): 10, 11 (21H2 or later)
- Mac: macOS 12, 13 (Apple Silicon is only available in 13)
- Linux CONFLEX: CentOS 7.6-7.9, RedHat EL 8.4, Ubuntu 22.04
- Specific surface energy calculation
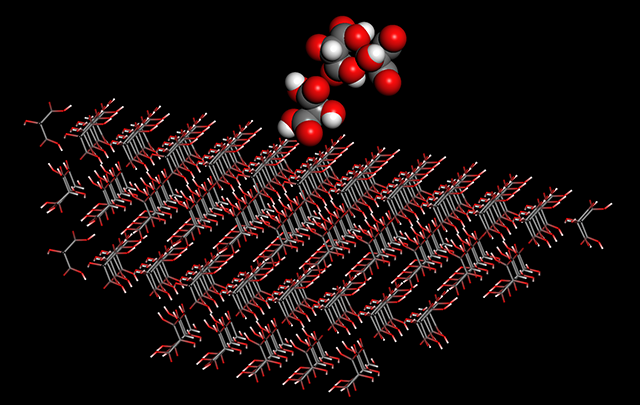
-
Calculation of specific surface energy is now available.
A crystal is a polyhedron consisting of multiple crystal planes and each specific surface energy of the planes are different. Since a crystal plane with large area in the polyhedron is considered to have a lower specific surface energy than other planes, the analysis using new feature is useful for study of crystal growth and morphology.
-
Calculation of specific surface energy is now available.
- Introducing the Distance Dependent Dielectric (DDD) method
- DDD method and its coefficient can now be introduced into the electrostatic term of force field. The default value of coefficient is 4.0.
- Hardware Requirements:
-
Newly supported platform from CONFLEX 9 Rev.B are highlighted in red.
Windows: 10, 11 (64bit)
Mac: macOS 10.15, 11, 12
Linux: CentOS 7.6-7.9, RedHat EL 8.4
- Enhancements to crystal structure optimization
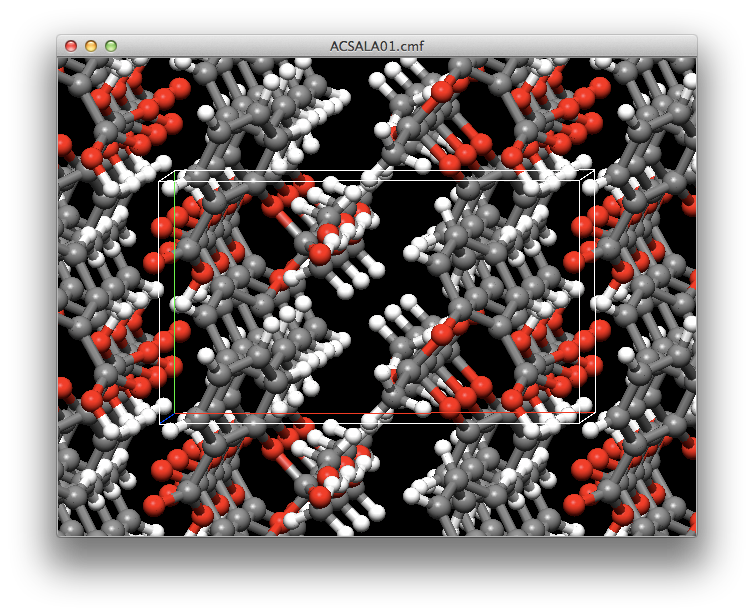
- The full diagonal Newton-Raphson method is now available for crystal structure optimization. This can significantly reduce the time required for crystal structure searches.
- Implementation the Ewald Method
- The Ewald method can now be used for electrostatic interaction calculations. The Ewald method can be used to calculate the electrostatic interaction over long distances with high speed and high accuracy.
- Calculation functions using AMBER force field
- The AMBER force field can now be used to optimize structures and search for conformers using input files for AMBER.
- Add option for GB/SA calculation
- Added an option to include only the electrostatic interaction term in the GB/SA calculation.
- Partial correction of MMFF94s parameters
- The N-H stretching parameters of the amide groups were returned to their original MMFF94s values.
- Operating System Requirements:
-
Newly supported platform from CONFLEX 9 Rev.A are highlighted in red.
Windows: 10 (32bit, 64bit)
Mac: macOS 10.14, 10.15
Linux: CentOS 7.6-7.9, 8.0-8.3
Linux・Interface: CentOS 7.6
-
Newly supported platform from CONFLEX 9 Rev.A are highlighted in red.
New Features in CONFLEX Interface 9
- Faster drawing of large molecule.
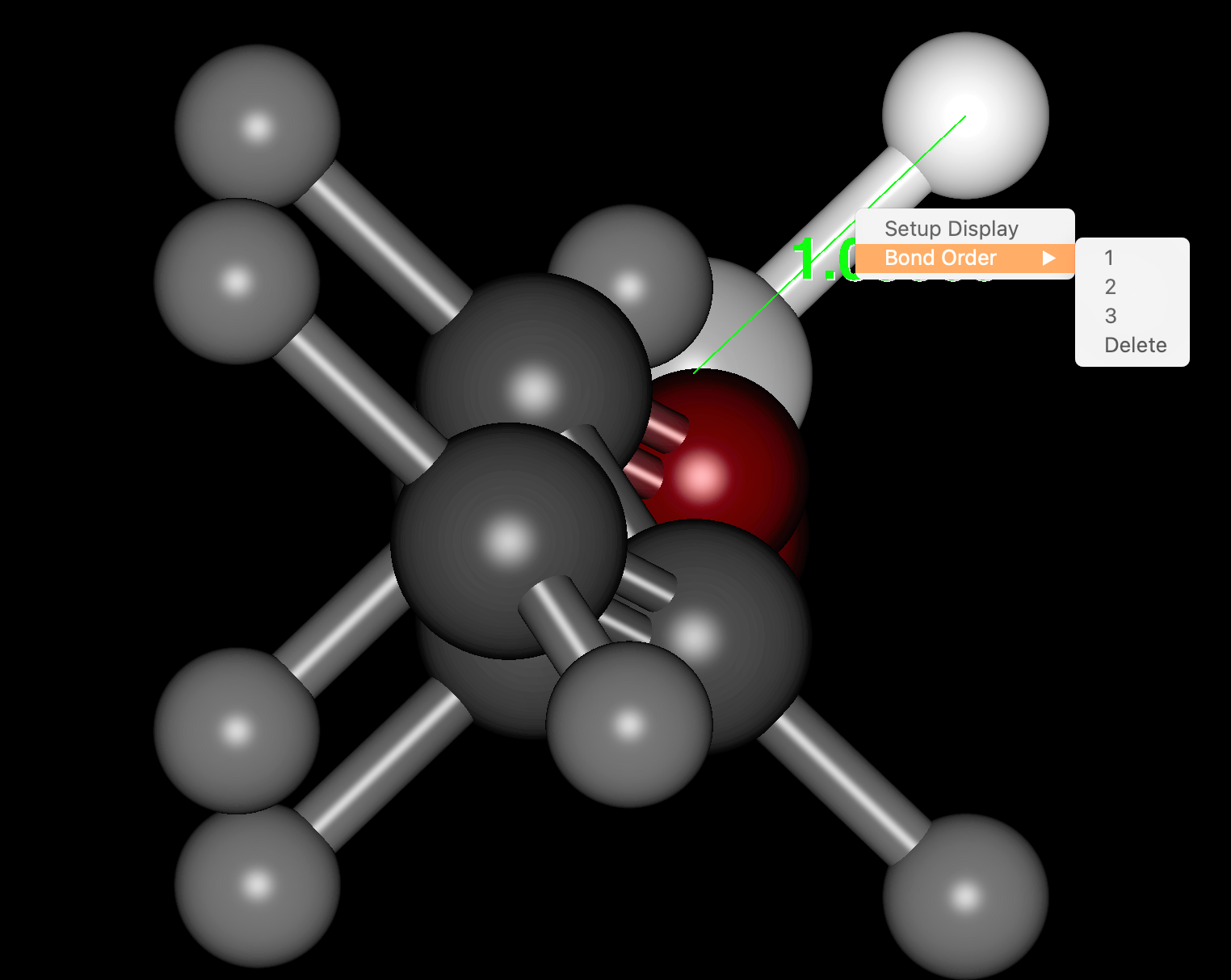
- Formal charges and Bond orders can now be edited in the molecule window (right mouse click).
- Files in cif/cmf format can now be saved.
- Support for cmf File containing multiple structures.
- CONFLEX Interface now support Apple Silicon (M1/M2).
- Operating System Requirements: Newly supported platform from CONFLEX 9 Rev.C are highlighted in red.
- Windows (64 bit): 10, 11 (21H2 or later)
- Mac: macOS 12, 13 (Apple Silicon is only available in 13)
- Linux Interface: RedHat EL 8.4, Ubuntu 22.04
-
New CONFLEX features are now supported.
- Specific surface energy calculation settings and results display
- Distance Dependent Dielectric Calculation Settings
- Improved summary display when CONFLEX BSO files is read.
-
Large files containing many conformations and crystal structures are now loaded in the background.
The waiting time before the first structure is displayed is reduced. -
Improved PDB and cif file reading routines.
Read errors have been reduced. - Operating System Requirements:
-
Newly supported platform from CONFLEX 9 Rev.B are highlighted in red.
Windows (64 bit): 10, 11 (21H2 or later)
macOS (x86_64): macOS 10.15, 11, 12
Linux: RedHat EL 8.4
-
Newly supported platform from CONFLEX 9 Rev.B are highlighted in red.
- The display format of molecules can now be partially changed.
- The display format of each area of a split screen can now be specified individually.
- An arbitrary value can now be specified for the display radius of an atom.
- Added support for OpenSSH, which is built into Windows 10 as a standard feature.
- You can now specify the line width when displaying wireframes.
-
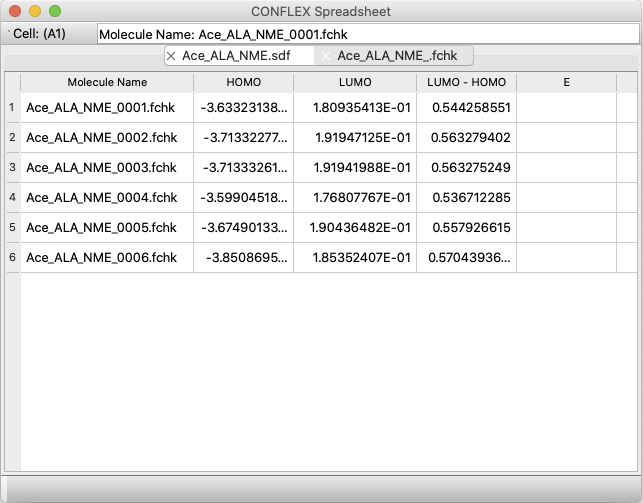
A new application, CONFLEX Spreadsheet, has been added.
- Reads a conformation file and automatically enters calculation properties in a tabular format.
- Read crystal search files and automatically enter calculation properties in tabular format.
- Read multiple Gaussian FChk files and automatically enter calculation properties in a tabular format.
- Copy and paste multiple cells
- Save in Microsoft Excel format
- Arithmetic functions for numbers in cells
CONFLEX 8 What's NEW
- Parameter setting for NMR 3J coupling constant calculation
- NMR 3J coupling constant calculation by using Karplus equation and setting parameters is available.
- It is possible to obtain 3J coupling constant between arbitrary atoms.
- Clustering of molecular crystal structures
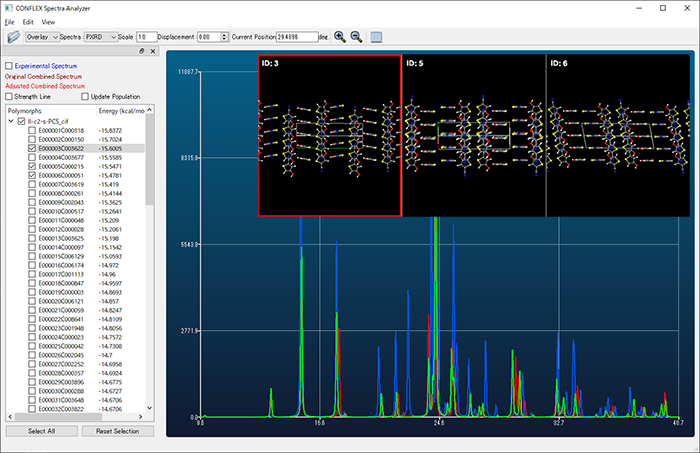
- Structures obtained by crystal search can be grouped based on similarity of powder X-ray diffraction pattern. Polymorphic structures close to a crystal structure can be easily found.
- Enhancement of crystal structure optimization
- Crystal structure can be optimized while fixing structure and position of specified molecule(s).
- Parameter customization for PPP calculation
- Hardware Requirements:
- Newly supported platform from CONFLEX 8 Rev.C are highlighted in red. Windows・CONFLEX&Interface: 8.1, 10
macOS・CONFLEX&Interface 10.13, 10.14
Linux・CONFLEX: CentOS 6.6-6.10, 7.2-7.6, 8.0
Linux・Interface: CentOS 7.2-7.6
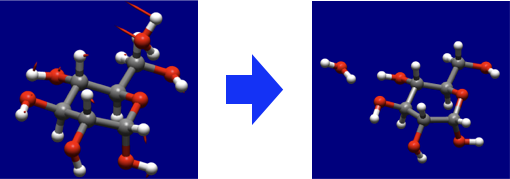
- Partially constrained geometry optimization and conformation search
- Geometry optimization with specifying atoms to be fixed is implemented.
- It is also possible to perform conformation search with that restriction.
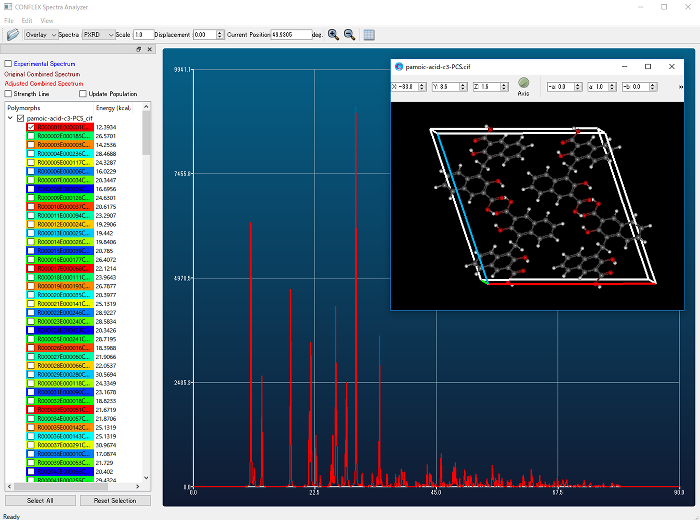
- Similarity calculation of powder X-ray diffraction patterns
- Crystal structures found by crystal search can be ranked by similarity between experimental and calculated powder X-ray diffraction patterns.
- Hardware Requirements:
-
Newly supported platform from CONFLEX 8 Rev.B are highlighted in red.
Windows・CONFLEX&Interface: 7, 8.1, 10
macOS・CONFLEX&Interface 10.12, 10.13
Linux・CONFLEX: CentOS 6.6-6.9, 7.2-7.5
Linux・Interface: CentOS CentOS 6.6, 7.2
-
Newly supported platform from CONFLEX 8 Rev.B are highlighted in red.
Windows・CONFLEX&Interface: 7, 8.1, 10
- Dynamic Reaction Coordinate (DRC)
- The molecular dynamics calculation using a vibrational frequencies and normal modes.
- DRC is applicable to simulation of coordination change for a system with multiple molecules, or conformation change of large molecule, etc...
-
Invoking external program (Gaussian 09, 16) for geometry optimization and conformation search.
- If Gaussian program is installed to a computer with CONFLEX program, a user can specify to invoke Gaussian program for geometry optimization.
- CONFLEX can now handle molecules without Force Field parameter(s) or electronic state which classical mechanics cannot treat.
- While using the same search algorithm of CONFLEX, you can evaluate the conformational isomers based on the energy values obtained by geometry optimization at the electronic structure calculation level.
-
Achieved high-efficient crystal optimization with OpenMP method (Parallel CONFLEX).
- Speed-up calculation of inter-molecular interaction energies with parallel treatment using OpenMP.
- With the introduction of the OpenMP code, the MPI parallel function for crystal structure optimization will be discontinued.
-
Achieved high-efficient crystal structure searching with hybrid OpenMP/MPI method.
- Implemented hybrid OpenMP/MPI method during crystal structure searching.
- CONFLEX 8 can parallelize crystal structure search with MPI method and crystal structure optimization with OpenMP method all together.
- Speed-up determining time of conformation to keep using their dihedral angles (CHECK=TORSION).
- Revised atomic weight used in calculation.
- B. Pfeiffer, K. Venkataramaniah, U. Czok, C. Scheidenberger, "Atomic mass compilation 2012", Atomic Data and Nuclear Data Tables, 2014, 100, 403-535.
- Hardware Requirements:
- Newly supported platform from CONFLEX 8 Rev.A are highlighted in red.
Windows: 7, 8.1, 10
Mac: macOS 10.11, 10.12
Linux: CentOS 6.6-6.9, 7.2-7.4
- Newly supported platform from CONFLEX 8 Rev.A are highlighted in red.
CONFLEX Interface 8 What's NEW
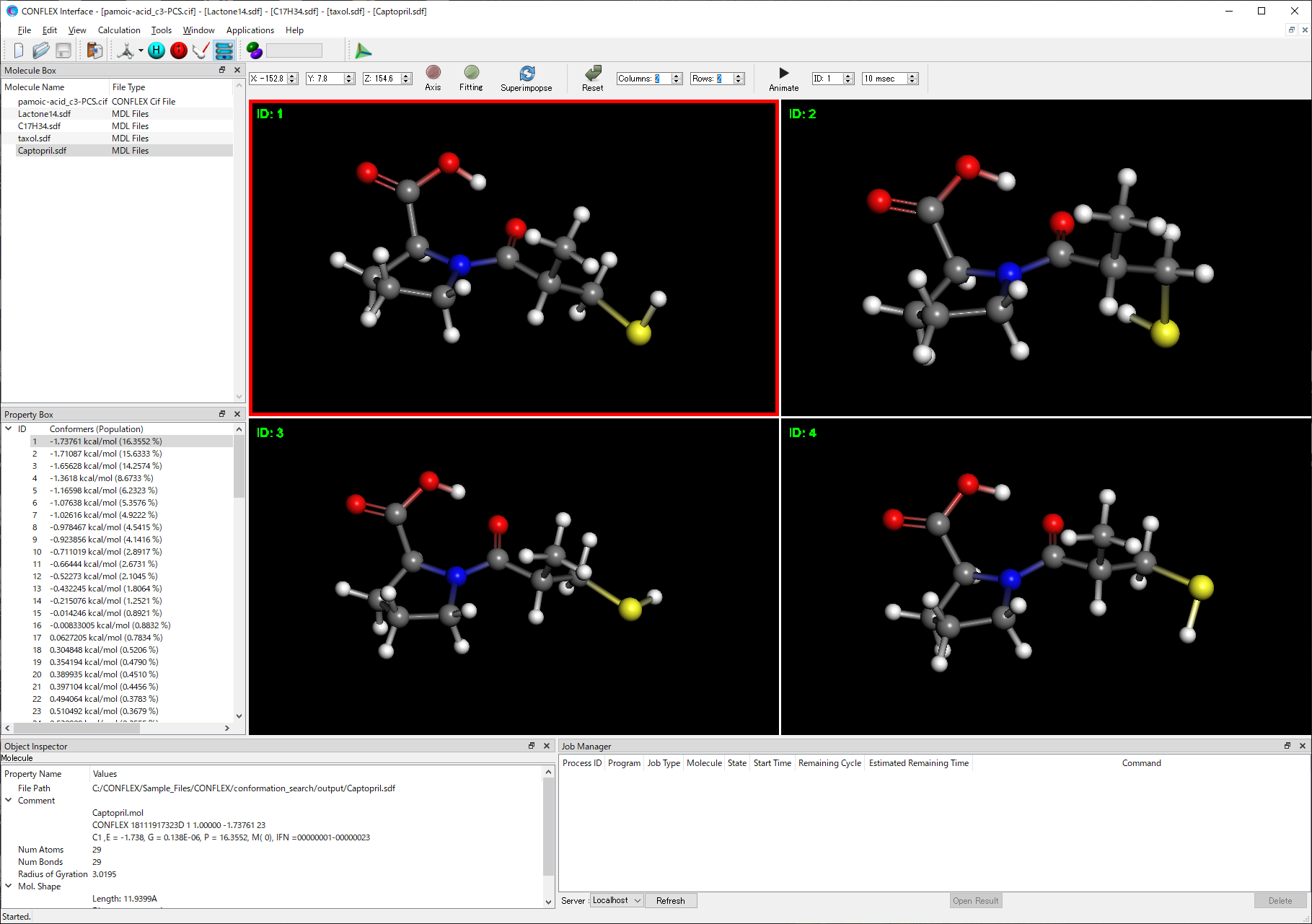
- Screen division
- Conformations or crystal structures are displayed in each divided screen.
- Superimposing
- Superimposing the multiple conformations based on specified atoms and displaying it.
- Sorting the list in property dialog
- Animation of the result of DRC calculation
- Acceleration of loading crystal search result file containing a large amount of structures
- Acceleration of graphic display by GPU (OpenGL 3.3 or later is required)
- Fit all conformers to minimize RMSd
- Superpose selected conformers
- Support PBS Pro for job scheduling
- Performance Enhancements
- Atom labeling is improved using GPU
- Spectra Analyzer Enhancements
- Optimizes a chart resolution according to zoom level
- Support X-ray diffraction spectrum
- Support to load experimental X-ray diffraction spectrum
- Labeling capability

- Enhancement of graphic display
- Enhanced performance using OpenGL Shading Language
- Animation display for vibrational mode
- Movie display for DRC calculation
- Display of crystal surface defined by h, k, l indices
- Line display of molecule
- (Mac) Support Retina display
- New Control Toolbar
- Toggle XYZ coordinate axis display
- Toggle unit box display of crystal
- Set the cell replication number to display
- Control button of animation (play, stop, frame-by-frame)
- Animation speed control
- Magnification of vibrational animation
- Transparency of molecular surface display
- Electron density to define the size of molecular surface
- New dialogs for calculation setup
- Host - Ligand Search
- DRC
- External program invoking
- OpenMP/MPI hybrid parallel
- Simplified crystal space selection page
- Enhancement for PDB file reading
- When you load PDB file containing amino acids or nucleic acids, Interface should add missing atoms.
- Geometry labels for atom distance, bond anble and torsion anble
- Watch List feature for geometry labels
- Label for atom distance, bond angle and torsion angle label between unbonded atoms
- Labels between molecules inside crystal
- Multiple labels display
- Changed the preference file path
- For security purpose, changed the preference file path from registry or inside Application package to the followings:
- Win: FOLDERID\_RoamingAppData/conflex.co.jp/CONFLEX.ini
- Unix: $HOME/.config/conflex.co.jp/CONFLEX.ini
- Mac: $HOME/.config/conflex.co.jp/CONFLEX.ini
- For security purpose, changed the preference file path from registry or inside Application package to the followings:
CONFLEX 7 What's NEW
- Calculate sum of interaction energies in hemispherical crystal
- CONFLEX can calculate sum of interaction energies in hemispherical crystal with respect to a molecule ‘in’ or ‘on’ a crystal plane specified by Miller indices.
- It is useful for the energy analysis regarding crystal plane such as dissolution,sublimation, and so on.
- Performance improvement for crystal structure search (Parallel CONFLEX)
- The method of parallel computation for the crystal structure search is implemented that geometry optimizations of crystal structures are distributed over multiple threads. (ONLY for Parallel CONFLEX)
- The performance of parallel computation for crystal structure search is vastly improved.
- Change the definition of crystal energy
- The definition of crystal energy is changed to improve the accuracy.
- Support for MOL2 file format
- CONFLEX now support MOL2 file format for direct CONFLEX calculation.
- Expanded torsion interaction
- CONFLEX can treat the higher order of torsion interaction term up to 6 dimensions.
- New keyword for fixed E-Z torsional configuration
- The keyword that fixed E-Z torsional configuration can be released is available during conformation search.
- Hardware Requirements: (Newly supported platform from Rev.D are highlighted in red.)
Windows: 7, 8.1, 10
Mac: OSX 10.9, 10.10, 10.11
Linux: CentOS 6.5, 6.6, 6.7, 6.8
- CONFLEX
- Host-Ligand Coordination Search
- The stable position and orientation of a molecule or an ionic species relative to other molecule(s) can be found by this function. It is useful for specifying the stable structure of dimers or complex molecules.
- Parallel CONFLEX for Windows
- MPI parallel calculation of conformation search and crystal structure optimization are available on Windows (Parallel CONFLEX is required).
- Host-Ligand Coordination Search
- Interface 7 Rev.C (Interface is now included in CONFLEX)
- User can change Search Limit setting inside Basic Settings dialog.
- User can setup Gaussian calculation and submit job to local machine.
- The new application Scheme Editor can modify settings of Basic settings dialog.
- The new application Spectra Analyzer can synthesize multiple spectra of conformers calculated by Gaussian application.
- User can rotate molecule by entering the desired angle value.
- Display molecule shape information inside the object inspector dialog.
- Display vibrational spectra in orientation anisotropic way.
- The local Parallel CONFLEX execution is supported on Windows multi-core machine.
- Platform LSF is supported as job scheduler.
- Hardware Requirements:
- CONFLEX 7C runs on:
- Windows: 7, 8.1 (32/64bit)
- Mac: OS X 10.8-10.10
- Linux: CentOS 6.1-6.6, 7.0, Ubuntu 12.04.3, 14.04
CONFLEX
- User modifiable force field parameters
- Crystal structure optimization includes external pressure conditions (only for triclinic, monoclinic, orthorhombic)
- Table view of conformation lists
- Enhanced isosurface view
- Supports the Platform LSF Job Scheduler
- Includes Scheme Editor which enables users to define calculation templates
- Plots spectra for IR, UV/Vis, UV/CD, NMR Peak Patterns
Interface
- Ability to display the distribution list (files with the extension ls1)
- Expanded the ability to create and display isoelectronic density surfaces.
- Support for Job Scheduler: Platform LSF
- Simple editing of calculation schemes
- Various spectrum display functions
(Vibrational spectrum, UV/Vis spectrum, UV/CD spectrum, proton NMR peak shape)
Hardware Requirements:
- CONFLEX 7B runs on:
- Windows: 7, 8.1 (32/64bit)
- Mac: OSX 10.6, 10.7, 10.8, 10.9
- Linux: CentOS 6.1-6.5, Ubuntu 12.04.3
- Crystal Structure Searching and optimization
- By inputting the molecular structure data and the symmetry of the space group, the CONFLEX automatically creates a crystal structure, performs structural optimization, and comprehensively calculates the crystal structure located at the energy minimum.
- For a series of optimized crystal structures, you can not only sort them in order of decreasing energy, but also sort them in order of proximity to pre-prepared powder diffraction data.
- Simulation of Powder Diffraction Patterns
- It calculates and outputs the powder diffraction data of crystal structure, and the element and wavelength of X-ray source can be changed.
- NEW CONFLEX Interface
- It is now possible to run jobs over a network. For example, you can run a calculation from a Windows PC to a Linux server. Also, the results obtained on the Linux server can be read directly from the Windows PC (GridEngine must be installed on the server).
- Integration with external programs is possible. For example, you can run Gaussian calculations over a network (GridEngine must be installed on the server).
- The following files can be read.
- MDL file:.mol .sdf
- CONFLEX出力 file:.bso .sdf
- Sybyl mol2 file
- PDB file
- Crystal structure file:.cif .cmf
- Gaussian Formatted Checkpoint file:.fchk
- GAMESS Log file
- Firefly Log file
- Cut & Paste from ChemDraw
- Can be launched from ChemOffice products
-
The ChemOffice version 13 now includes the CONFLEX interface.
If you purchase ChemBioOffice Ultra v13, ChemOffice Pro v13, or ChemBio3D Ultra v13, you can run CONFLEX calculations directly.